This page was created as an undergraduate assignment for Davidson College.
Review Paper Project
Integrin a3b1 (CD 49c/29) Is a Cellular Receptor for Kaposi’s
Sarcoma-Associated Herpesvirus (KSHV/HHV-8) Entry into the Target Cells
Shaw M. Akula, Naranatt P. Pramod, Fu-Zhang Wang, and Bala Chandran. 2002. Cell.
108: 407-419.
Reviewed by Danielle Hyun-jin Choi
The researchers of this article studied the role of integrins to act as receptors
for the herpesvirus envelope glycoprotein B (gB). Human herpesvirus-8 (HHV-8)
is associated with Kaposi’s Sarcoma (KS), most commonly seen in AIDS patients.
The patients develop malignant skin tumor that results in multifocal purplish
colored sores that eventually form nodules, small lumps of hard swelling tissues,
even within the lungs and intestines.3
HHV-8 envelope protein contains the RGD motif with which integrins interact.
To study the binding of HHV-8’s RGD to integrins, the researchers used
artificial RGD peptides and antibodies against RGD-binding integrins to detect
inhibition of HHV-8 infection. The artificial RGD protein constructs, the antibodies
against RGD amino acid, and the soluble a3b1 integrin significantly blocked
infection.
The researchers also reasoned that if more RGD-dependent integrins were expressed
on the cell membranes, the more receptors there would be for HHV-8, and more
infection would be obtained. And they did observe increased infection in Chinese
hamster ovary cells, when human a3 integrin was artificially expressed on the
cell surface.
Immunoprecipitaion method was used to study the binding of anti-gB antibody
to specific chains of the integrin proteins. Further investigation involved
the detection of integrin-dependent focal adhesion kinase (FAK) activation with
soluble a3b1 integrins and anti-gB antibodies. Interestingly, the HHV-8 infection
inducing factors seemed to phosphorylate FAK.
Fig 1. RGD peptides inhibit HHV-8 infection.
Green Fluorescent Protein (GFP-HHV-8) and antibodies against HHV-8 ORF73 were
used to detect the level of infection. Control peptides did not include RGD
amino acids, while the treatment peptides contained RGD. Panels A and D were
treated with control peptides and were mock infected, so no signal of infection
was detected. In panels B and E, the cells were treated with the control peptides,
and were infected with GFP-HHV-8. As expected, I see signals for infection.
The interesting data come in C and F: when the cells were treated with RGD peptides
and with GFP-HHV-8, I see relatively less signal than shown in B and F. I agree
with the authors that the RGD peptides inhibited GFP-HHV-8 infection. I also
liked the idea of screening for the HHV-8 ORF73 proteins, because the only source
of those proteins is HHV-8, and allowed the researchers to measure infection
by non-GFP HHV-8, since the “GFP-HHV-8” actually contained both wild-type
and recombinant viruses.
Fig 2. Antibodies against RGD peptides and other integrin ligands also inhibit
HHV-8 infection.
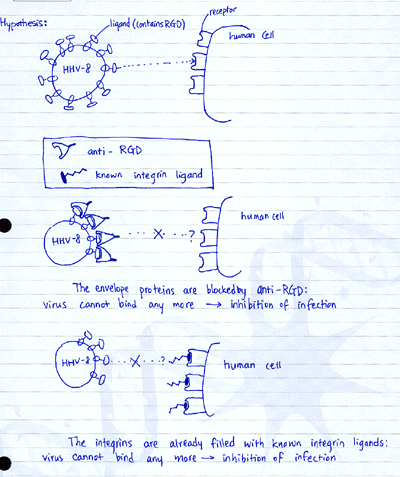
The researchers hypothesized that if integrins are acting as receptors and HHV-8
envelope proteins as ligands, blocking the ligands should inhibit binding to
the receptors. Also, if the receptors (integrins) have already bound to other
ligands, the receptors are blocked, so that the virus envelope protein cannot
bind to integrins. (See diagram below)
Indeed, the data shown here seem to support that hypothesis. Graph A shows a
case for “blocking the receptor” by RGD peptides. Compared to the
non-RGD peptide, higher level of inhibition was observed for RGD peptide treatment.
Graph B shows a case for “coating the ligand” by antibodies against
virus envelope proteins. Since antibodies against RGD peptide coated the ligands
around the virus, higher inhibition was observed compared to non-specific antibodies.
Graph C is another case for “blocking the receptor” mechanism of inhibition
by known integrin ligands (fibronectin), compard to a variety of other ligands.
Fig 3. Soluble a3b1 integrin inhibits HHV-8 infection.
Now the researchers have moved on to identify which integrins are involved in
HHV-8 gB binding and infection. They have used a variety of anti-integrin antibodies,
for some a units and b units separately, and some in combinations. They measured
% inhibition of GFP-HHV-8 infection, and found that a3 was the most effectively
inhibited integrin, and then b1 and a2b1 integrins were similarly inhibited
in two different types of cells (Graph A). Curiously, they did not include a3b1,
which they suggested to be the receptor for HHV-8 gB. They show that inhibition
by anti-integrin antibodies was dose-dependent (Graph B). I think one of the
most important figures is Graph C, because the graph depicts the specificity
of ligand-receptor interaction for HHV-8 gB and a3b1 integrin. See the diagram
below for why soluble a3b1 integrin would inhibit infection.
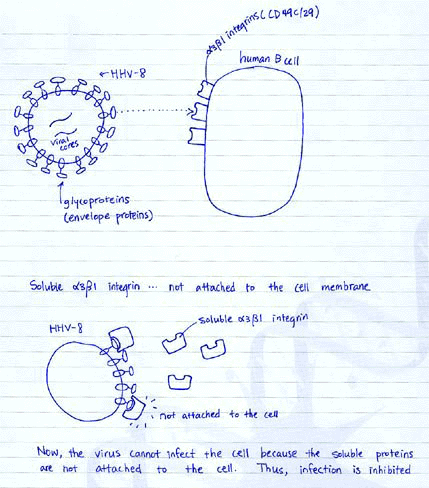
Fig 4. Determining the relative abundance of a3b1 integrin.
Flow cytometric analysis was used. The higher the integrin expression level
was, the more intense the fluorescence signal was detected for this FACS data.
A is the control (no integrin detected), B for avb3 integrin expression, C for
a3, and D for b1. The data suggests that significantly more a3 and b1 integrins
were expressed and detected, compared to the control expression level. Table
E shows the relative abundance of target integrin expression in different types
of cells with Mean Fluorescence Intensity value in paranthesis. I note that
the relative abundance of a3 and b1 is well over 90% for BJAB and HFF cells.
Fig 5. Expression of human a3 integrin in hamster ovary cells (CHO-B2) increase
HHV-8 infection.
As the percentage of human a3 expression increased in CHO-B2 clone D5 cells
(2) and even more in CHO-B2 clone B3 cells (3) compared to the control (1),
the intensity of fluorescence increased, indicating an increase in GFP-HHV-8
infection in those cells. In Panel B, more signal for GFP-HHV-8 infection is
observed from B3 cells (2 and 4), than D5 cells (1 and 3). This panel seems
to be missing the control. The researchers should have shown us a GFP detection
of cells that were not infected with GFP-HHV-8. Also, panel B seems redundant
since the differences in fluoresence intensity for expression of GFP-HHV-8 in
D5 virsus B3 cells were already shown in panel A (2 and 3). Panel C shows the
increased susceptibility to HHV-8 infection as a function of expression of human
a3 integrins. The middle bars represent inhibited infectivity due to the antibodies
added, thus coating the virus envelope proteins with antibodies to inhibit infectivity,
and this procedure proves that the integrins of interest and GFP-HHV-8 are the
key players at work. Anti-b4 cannot coat the virus proteins, so no inhibition
is observed (the right most bars). It seems fairly convincing that the increase
in expression of human a3 integrins resulted in the increase in infectivity.
But I think the mechanism by which the hamster b1 and human a3 integrins interact
was not addressed clearly, which is critical for the explanation of increase
in infectivity.
Fig 6. Immunoprecipitation of the virus a3 and b1 complexes with anti-gB antibodies.
The main point of this figure is to show that the anti-gB (HHV-8 glycoprotein
B) antibodies specifically immunoprecipitated a 150/110 kDa heterodimer protein,
which was identified as a3 and b1 (lanes 7, 8, 10, 11). However, anti-gB antibodies
did not immunoprecipitate a1 chain of the integrin. Heparinase was added to
prevent undesirable interaction between HHV-8 and the ubiquitous cell surface
haparin sulate-like molecule. Their claim that HHV-8 probably recognizes a specific
conformation of a3b1 only from this data seems bold to me. Further study seems
to be required to see the exact conformational interaction between HHV-8 gB
and the a3, b1 chains of the integrins. Panel B shows that only heparin blocked
binding of HHV-8 to HFF cells, while other RGD peptides, antibodies to RGD gB
and other a3b1 associated proteins did not. The failure to inhibit binding suggests
that a3b1 integrin interacts with HHV-8 at a postattachment step of infection.
Panel B study seems too open-ended to me.
Fig 7. Panel A shows the change in distributions of Focal Adhesion Kinase in
the cell after HHV-8 infection or after lysophosphatidic acid (LPA) treatment.
For FAK to change distributions by colocalizing with vinculin, FAK must be phosphorylated
first. After 5 minutes of HHV-8 infection, FAK showed colocalization with vinculin
in panel A. It seems convincing enough that FAK was phosphorylated due to HHV-8
infection and changed distributions. For panel B, the gradual increase in the
intensity of the bands from lane 3 to 5, suggest that longer exposure to HHV-8
induced more phosphorylation of FAK. (The bands indicate that anti-phospho-FAK
antibodies bound to the phosphorylated form of FAK.) For C, soluble a3b1 integrins
were added in decreasing amount from lanes 2 to 4, and the increase in the band
intensities sugguest that the soluble integrins inhibited phosphorylation of
FAK. For D, anti-gB antibodies were added in decreasing amount from lanes 2
to 4, and the increase in the band intensities suggest that the anti-gB blocked
phosphorylation of FAK. These data seem fairly convincing and clear to me that
a3b1 integrins play a direct role in phosphorylation of FAK.
Overall, the authors of this study have indeed convinced me that a3b1 integrins
are acting as receptors to HHV-8 glycoprotein B. Their hypothesis that a3b1
integrins are receptors that bind to HHV-8 gB to permit entry of the virus into
the host cells seems congruent with the data they have given. There are some
loose strings I have noted in the figures explanations, but this paper is fairly
straight forward.
The identification of a3b1 integrins as receptors to HHV-8 opens up many future
study directions. First of all, since present study was done in vitro, only
in test tubes, it would be sensible to study the in vivo roles of these receptors
in HHV-8. Since GFP expression does not destroy the cells or inturrupt with
the cell functions (at least there have not been evidence that GFP inturrupts
with the life of the cells), we will be able to detect precisely which cells
in the organism have been infected by HHV-8 with GFP constructs, when the infection
occurs at what periods of time in the organism’s life, and relative abundance
of expression in each cell type. The researchers have already mentioned that
HHV-8 infection have been detected in vivo in human B cells, endothelial cells,
epithelial cells and others in the introduction. So now, with the knowledge
such as that soluble a3b1 integrins can block HHV-8 infection, that a3b1 integrin
ligands and anti-RGD antibodies can also inhibit infection, we can now investigate
how effective the inhibition is in vivo. This type of study may lead to discovery
of vaccines that are effective in preventing or treating HHV-8 infections. Since
HHV-8 has been known to be associated with Kaposi’s Sarcoma, such vaccine
may be revolutionary in treating or preventing KS. Even if the inhibition turns
out to be far less effective in vivo compared to in vitro, such results may
lead to discovery of some other proteins present in the cells (but absent in
test tubes) or possibilities for other mechanisms involved in this receptor-ligand
scenario.
Secondly, the researchers noted in their discussions that the lack of complete
inhibition of infection by soluble a3b1 integrins suggest that a3b1 integrins
are not the only receptor proteins. They may be able to utilize the gene that
encodes for a3b1 integrins (I assume they already have this information, since
plasmid constructs were used to express human a3 integrins in hamster cells),
to search for homologues or similarities in other organisms, especially those
known to be infected by HHV-8. Since we cannot use humans as our exprimental
subjects to see if they develop KS or not, if the researchers find very similar
mechanism and integrins at work in mice or hamsters, such discoveries may enable
more in vivo experiments. Such in vivo experiments may help to understand how
HHV-8 facilitate KS pathogenesis.
Lastly, the enzyme cascade involving focal adhesion kinase (FAK) can be further investigated. Although we have learned that blocking HHV-8 infection also blocks phosphorylation of FAK, we still don’t know what this phosphorylation exactly acheives in the cells. The antibodies used to detect phosphorylation in Figure 7 was anti-phospho-FAK antibodies, specifically designed to detect phosphorylation of only the FAK. Since enzyme cascades usually involve multiple phosphorylations in different proteins involved in the mechanism, I wonder if we can screen for any other proteins in the cells that are usually not phosphorylated, but specially phosphorylated due to the HHV-8 infection. I think that if such phosphorylated proteins are indeed found other than FAK, then may be the elusive enzyme cascade may be not too far from understanding.
1Purves, W.K., Orians, G.H., Heller, H.C., Sadava, D. (1998).
Life. The Science of Biology. 285.
2Purves, 424.
3On-lineMedical Dictionary, Kaposi Sarcoma, 1997. http://cancerweb.ncl.ac.uk/cgi-bin/omd?query=Kaposi+Sarcoma

![]()
![]()
![]()