New England Journal of Medicine. Volume 35. Number 8. 1996
OVEREXPRESSION OF AN OSTEOGENIC MORPHOGEN IN FIBRODYSPLASIA OSSIFICANS PROGRESSIVA
ADAM B. SHAFRITZ,
B.A., EILEEN M. SHORE,
PH.D., FRANCIS H.
GANNON, M.D., MICHAEL
A. ZASLOFF, M.D., PH.D.,
REBECCA TAUB, M.D.,
MAXIMILIAN MUENKE,
M.D., AND FREDERICK S. KAPLAN,
M.D.
ABSTRACT
Background Fibrodysplasia ossificans progressiva is
a heritable disorder of connective tissue characterized by congenital
malformation of the great toes and postnatal formation of ectopic
bone. Although the disorder was first described more than 300
years ago, the genetic defect and pathophysiology remain unknown.
Bone morphogenetic proteins are potent bone-inducing morphogens
that participate in the developmental organization of the skeleton,
and increased production of one or more of these proteins has
been proposed as the cause of fibrodysplasia ossificans progressive
Methods We studied lymphoblastoid cell lines established
from peripheral-blood mononuclear cells of patients with fibrodysplasia
ossificans progressiva and fibroblast-like cell lines derived
from lesional and nonlesional tissue. We used Northern blot analysis
and ribonuclease protection assays to measure the expression of
messenger RNA (mRNA) of bone morphogenetic proteins 1 to 7 and
immunohistochemical analysis to examine protein expression.
Results Among the bone morphogenetic proteins and mRNAs
examined, only bone morphogenetic protein 4 and its mRNA were
present in increased levels in cells derived from an early fibroproliferative
lesion in a patient with fibrodysplasia ossificans progressiva.
Bone morphogenetic protein 4 mRNA was expressed in lymphoblastoid
cell lines from 26 of 32 patients with fibrodysplasia ossificans
progressiva but from only 1 of 12 normal subjects (P<0.001).
Bone morphogenetic protein 4 and its mRNA were detected in the
lymphoblastoid cell lines from a man with fibrodysplasia ossificans
progressiva and his three affected children (two girls and a boy),
but not from the children's unaffected mother. No other bone morphogenetic
proteins were detected.
Conclusions Overexpression of a potent boneinducing morphogen
(bone morphogenetic protein 4) in lymphocytes is associated with
the disabling ectopic osteogenesis of fibrodysplasia ossificans
progressiva.
(N Engl J Med 1996;335:555-61.) ©1996, Massachusetts
Medical Society.
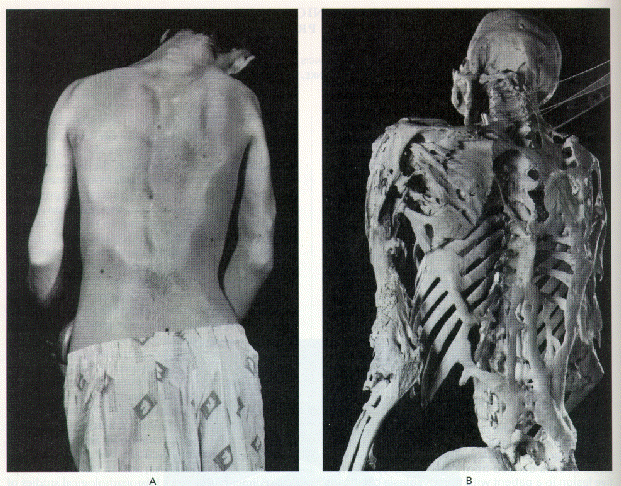
Figure 1. Clinical
Appearance and Skeleton of a Man with Fibrodysplasia Ossificans
Progressiva.
The rigid posture in this 25-year-old man with fibrodysplasia
ossificans progressiva was due to ankylosis of the spine, shoulders,
and elbows. He died of pneumonia at the age of 40 years. Plates
and ribbons of ectopic bone contour the skin over the back and
arms (Left Panel), and can be seen directly on the skeleton (Right
Panel). Courtesy of the Mütter Museum, College of Physicians
of Philadelphia.
Studies to identify the cause of fibrodysplasia ossificans progressiva are currently focused on the candidate-gene approach,10 since karyotypic abnormalities have not been detected in patients with the disorder3 and lesional tissue is not readily available for study.7 Definitive linkage analysis is not possible, since only three small families with inheritance of fibrodysplasia ossificans progressiva have been identified worldwide, 2,3,11 and the molecular defect is not known. In this article, we provide evidence that a potent bone-inducing morphogen, bone morphogenetic protein 4, is overexpressed in the lymphocytes of patients with fibrodysplasia ossificans progressiva.
METHODS
Patients and Cell Lines
From 1990 to 1995, 62 patients with fibrodysplasia ossificans
progressiva were referred to the fibrodysplasia ossificans progressiva
working group at the University of Pennsylvania. Lymphoblastoid
cell lines were established from peripheral-blood mononuclear
cells in 32 patients and 12 normal subjects by transformation
of the cells by Epstein-Barr virus.12
The protocols were approved by the institutional review boards
of the Childrens's Hospital of Philadelphia and the University
of Pennsylvania, and informed consent was obtained from all patients
or their parents.
Primary cell lines were established from pre-osseous lesional tissue from a 15-year-old boy with fibrodysplasia ossificans progressiva, after resection of a lesion affecting the muscles of mastication; uninvolved skin and subcutaneous tissue from an 8-year-old girl with fibrodysplasia ossificans progressiva, removed at the time of an emergency craniotomy for resection of a craniopharyngioma, an apparently unrelated condition); skin and subcutaneous tissue from a normal infant; paraspinal muscle from a normal 4-year-old girl; and temporalis muscle from three normal children, ages 1, 6, and 14 years, who had undergone unrelated neurosurgical procedures. The skin fibroblast cells from the normal infant were used as a negative control, and the U-2 OS osteosarcoma cell line (American Type Culture Collection) was used as a positive control for the expression of bone morphogenetic proteins.13
Cell Culture
Primary cultures of fibroblast-like cells were established in
Dulbecco's modified Eagle's medium containing 2.5 µg of
amphotericin B (Fungizone) per milliliter, 50 µg of gentamicin
per milliliter, and 15 percent fetal-calf serum. The U-2 OS osteosarcoma
cells were grown in McCoy's 5A medium and 2.5 µg of amphok
tericin B per milliliter, 50 µg of gentamicin per milliliter,
and 10 percent fetal-calf serum. Lymphoblastoid cell lines were
grown in RPMI 1640 medium containing 100 units of penicillin per
milliliter, 100 µg of streptomycin per milliliter, 0.25
µg of amphotericin B per milliliter, and 15 percent fetal-calf
serum. All Stissue-culture reagents were obtained from Life Technologies
(Gaithersburg, Md.). The control and fibrodysplasia ossificans
progressiva lymphoblastoid cell lines were coded and run together
in assays, and the results were read in a blinded fashion.
Preparation of RNA
Total RNA was isolated from lymphoblastoid cell lines and primary
cells in culture by extraction with guanidine isothiocyanate and
phenol-chloroform-isoamyl alcohol. 3,4
Poly( A)+ RNA us prepared from
total cellular RNA with oligo(dT) cellulose (5' Prime-3' Prime,
Boulder, Colo.).
Complementary DNA Clones
Plasmid clones containing human complementary DNA (cDNA) inserts
from the following genes were used as templates for labeled-probe
synthesis in Northern blot analyses and ribonuclease protection
assays: collagen types I and II, osteocalcin, and alkaline phosphatase
(liver-bone-kidney type) (used to establish the cell phenotype);
c-fos, c-jun, c-jun B, c-jun D, and
transforming growth factor ß1 (TGF-ß1) (genes involved
in the early cellular response and terminal differentiation pathways
of chondro-osseous development)15,l6;
MSX-2 (a homeobox gene induced during osteogenesis)17; and bone morphogenetic proteins 1
to 7 and growth-differentiation factor 5 (members of the family
of bone morphogenetic proteins).15
The expression of glyceraldehyde-3-phosphate dehydrogenase was
used as an internal standard.
Northern Blot Analysis
With the use of standard methods poly(A)+
RNAs (4 µg) from cultured human cells were electrophoresed
through 0.8 percent agarose formaldehyde denaturing gels, transferred
to nylon membranes (MagnaGraph, MSI, Westborough, Mass.), and
prehybridized with 5X saline sodium citrate (SSC; 1X SSC is 0.15
M sodium chloride and 0.015 M sodium citrate per liter), 50 percent
formamide, 6X Denhardt's solution (1X Denhardt's solution is 0.02
percent Ficoll, 0.02 percent polyvinylpyrrolidone, and 0.02 percent
bovine serum albumin), 300 mg of denatured salmon-sperm DNA per
milliliter of solution, and 0.1 percent sodium dodecyl sulfate
at 42° C. Probe cDNAs were labeled with [a-32P]deoxycytidine triphosphate by random-primer
extension. Membranes were hybridized with 2 x 106
cpm of probe per milliliter in 50 percent formamide, 5X SSC, 3X
Denhardt's solution, 150 mg of denatured salmon-sperm DNA per
milliliter, and 0.1 percent sodium dodecyl sulfate. The blots
were washed under conditions designed to remove nonspecific hybridization
and were then subjected to autoradiography. To remove labeled
cDNA for reprobing, the hybridized blots were incubated in 60
percent formamide, 50 mM TRIS-hydrochloric acid (pH 8.0), and
1 percent sodium dodecyl sulfate for one hour at 75° C.
Ribonuclease Protection Assays
Antisense RNA probes were synthesized by in vitro transcription
(MAXIscript, Ambion, Austin, Tex.), and full-length transcripts
were purified from a 5 percent acrylamide, 8 M urea gel with 0.5
M ammonium acetate, 1 mM EDTA, and 0.1 percent sodium dodecyl
sulfate. For each ribonuclease protection assay, 150,000 cpm of
[32P]-labeled antisense RNA
was used according to recommended protocols (RPA II, Ambion).
The samples were denatured and electrophoresed through a 5 percent
acrylamide, 8 M urea gel. The dried gel was subjected to autoradiography
and then exposed to a phosphor screen. Messenger RNAs (mRNAs)
were quantitated with either a Computing densitometer (model 300A,
Molecular Dynamics, Sunnyvale, Calif.) or a PhosphorImager (Molecular
Dynamics).
Immunohistochemical Analysis
Lymphoblastoid cells from 10 patients with fibrodysplasia ossificans
progressiva and 10 normal subjects were applied to uncoated glass
slides. The cells were stained according to standard immunoperoxidase
protocols with antibody against bone morphogenetic proteins 2
and 4 (1:3000 dilution), counterstained with Wright's stain, and
examined under light microscopy. Slides of an osteosarcoma (positive
control) and liver tissue (negative control) were stained simultaneously
(data not shown).
RESULTS
Pattern of mRNA Expression in Cells from a Fibrodysplasia Ossificans Progressiva Lesion
The expression of mRNAs of genes related to bone and cartilage development in pre-osseous lesional tissue from a patient with fibrodysplasia ossificans progressiva was evaluated by Northern blot analysis. The lesional cells were morphologically similar to skin fibroblasts. The pattern of gene expression of the two types of cells was similar, with the exception of mRNA of bone morphogenetic protein 4, which was detected in the lesional cells from a patient with fibrodysplasia ossificans progressiva but not in skin fibroblasts from normal subjects or the patient (Table 1). Bone morphogenetic protein 4 was the only TGF-ß-like bone morphogenetic protein expressed in the early lesional cells. There was no detectable expression of bone morphogenetic protein 2, 3, 5, 6, or 7; collagen type II; osteocalcin; or MSX-2 in either lesional cells or normal skin fibroblasts (Table 1). Type I collagen mRNA was expressed at higher levels by skin fibroblasts than by lesional cells from patients with fibrodysplasia ossificans progressiva. The levels of expression of alkaline phosphatase, c-fos ,c-jun, c-jun B, c-jun D, TGF-ß1, and growth-differentiation factor 5 were similar in lesional cells and normal skin fibroblasts.
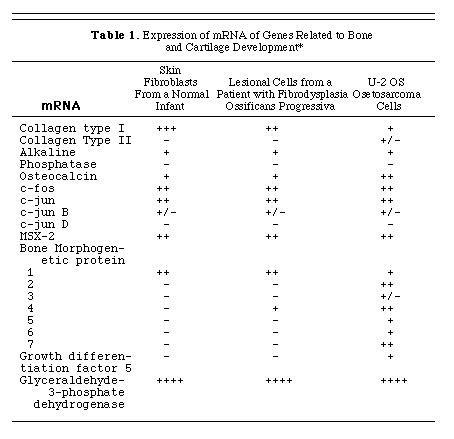
* The + + + + symbol indicates an intense signal after 2 hours of exposure of a hybridized Northern blot to X-ray film (see the Methods section); + + + indicates an intense signal after 12 hours of exposure; + + indicates a strong signal within 24 hours; + indicates a signal that was easily seen within a 3-day period; and +/- indicates a faint signal at 7 days. The - symbol indicates that no signal was detected after seven days of exposure at -70° C with two intensifier screens.
Expression of Bone Morphogenetic Protein mRNA in Nonlesional and Lesional Cell Lines from Patients with Fibrodysplasia Ossificans Progressiva
RNAs from cell lines established from lesional and nonlesional sites from patients with fibrodysplasia ossificans progressiva and from the same sites in normal subjects matched for age and sex were evaluated for the expression of bone morphogenetic proteins 1 to 7 by ribonuclease protection assays (Fig. 2). Bone morphogenetic protein 1 mRNA was expressed in all cell lines examined, although the level of expression varied. Bone morphogenetic protein 2 mRNA, which was not detected by Northern blot analysis (Table 1), was expressed at very low levels in all the cell lines (data not shown). The expression of bone morphogenetic protein 4 mRNA was barely detectable in nonlesional cells from a patient with fibrodysplasia ossificans progressiva (Fig. 2, lane 3) and in normal skin fibroblasts (Fig. 2, lane 4). All the muscle-cell lines (Fig. 2, lanes 5, 6, 7, and 8) from normal subjects expressed bone morphogenetic protein 4 mRNA, but the highest level of expression was in the cell line from early lesional tissue from a patient with fibrodysplasia ossificans progressiva (Fig. 2, lane 2); in this cell line the level of expression was approximately 12 times greater than in cell lines derived from normal muscle or fascia.
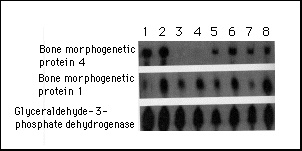
Figure 2. Expression
of mRNA of Bone Morphogenetic Protein 4.
Total RNA was analyzed for mRNA of bone morphogenetic proteins
4 and 1 by a ribonuclease protection assay with the probes indicated
to the left of each row. The expression of glyceraldehyde-3-phosphate
dehydrogenase mRNA served as an internal standard. RNA samples
were prepared from U-2 OS osteosarcoma cells (positive control)
(lane 1); pre-osseous lesional cells from a patient with fibrodysplasia
ossificans progressiva (lane 2); skin and subcutaneous cells from
another patient with fibrodysplasia ossificans progressiva (lane
3); skin and subcutaneous tissue from a normal subject (lane 4);
temporalis muscle, tendon, and fascia from normal subjects who
were 14 years old (lane 5), 6 years old (lane 6), and 1 year old
(lane 7); and cervical paraspinal muscle, tendon, and fascia from
a normal 4-year-old subject (lane 8).
Expression of Bone Morphogenetic Protein mRNA in Lymphoblastoid
Cell Lines from Patients with Fibrodysplasia Ossificans Progressiva
In order to determine whether dysregulation of the expression
of the bone morphogenetic protein 4 gene was a generalized feature
of fibrodysplasia ossificans progressiva, RNAs isolated from lymphoblastoid
cell lines from 32 patients with fibrodysplasia ossificans progressiva
and 12 normal subjects were examined for the expression of mRNAs
of bone morphogenetic proteins 1 to 7 by a ribonuclease protection
assay. Only the expression of bone morphogenetic protein 4 mRNA
differed between lymphoblastoid cell lines derived from patients
with fibrodysplasia ossificans progressiva and normal subjects
(Fig. 3). Lymphoblastoid cell lines from 26 of the 32 patients
with fibrodysplasia ossificans progressiva expressed bone morphogenetic
protein 4 mRNA, as compared with only 1 of 12 normal subjects
(P<0.001). No other bone morphogenetic proteins were expressed
by either group of cells. There was no apparent correlation between
age, sex, or severity of disease activity and the presence or
magnitude of expression of bone morphogenetic protein 4 mRNA.
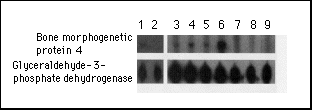
Figure 3. Expression
of Bone Morphogenetic Protein 4 mRNA in Immortalized Lymphoblastoid
Cell Lines from Patients with Fibrodysplasia Ossificans Progressiva
and Normal Subjects.
RNAs from lymphoblastoid cell lines were analyzed for the expression
of mRNA of bone morphogenetic protein 4 by a ribonuclease protection
assay. The expression of glyceraldehyde-3-phosphate dehydrogenase
mRNA served as an internal standard. Lanes 1, 3, 4, 5, and 6 show
RNA from patients with fibrodysplasia ossificans progressiva;
lanes 2, 7, 8, and 9 show RNA from normal subjects. The samples
from patients with fibrodysplasia ossificans progressiva are representative
of the range of detectable bone morphogenetic protein 4 mRNA.
Quantitation by densitometry (with correction for film background
levels and glyceraldehyde-3-phosphate dehydrogenase levels) yielded
values of zero for samples negative for bone morphogenetic protein
4 (11 of 12 normal subjects and 6 of 32 patients with fibrodysplasia
ossificans progressiva). The one normal subject scored as positive
for bone morphogenetic protein 4 mRNA had a value equal to the
lowest positive value measured for any patient with fibrodysplasia
ossificans progressiva. The range of expression of bone morphogenetic
protein 4 mRNA in the patients varied by a factor of about 20
from the weakest positive signal (lane 3) to the strongest positive
signal (lane 6).
In order to study the expression of bone morphogenetic protein 4 mRNA in lymphoblastoid cell lines derived from a kindred with fibrodysplasia ossificans progressiva, a family with genetic transmission of the disorder from parent to offspring3 was evaluated by a ribonuclease protection assay. Bone morphogenetic protein 4 mRNA was detected in cells derived from all four family members with fibrodysplasia ossificans progressiva - the father, two daughters, and a son - but not in cells from the unaffected mother (Fig. 4).
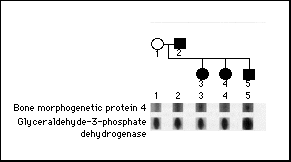
Figure 4. Expression
of Bone Morphogenetic Protein 4 mRNA in a Family with Fibrodysplasia
Ossificans Progressiva.
The pedigree of a family with genetic transmission of fibrodysplasia
ossificans progressiva from parent to offspring is shown. Circles
denote female family members, squares male family members, and
solid symbols affected family members. The father, both daughters,
and the son all have fibrodysplasia ossificans progressiva. The
mother is unaffected. RNA from lymphoblastoid cell lines established
from each subject was examined for bone morphogenetic protein
4 mRNA by a ribonuclease protection assay. The unaffected mother's
lymphoblastoid cells did not express bone morphogenetic protein
4 mRNA, whereas lymphoblastoid cells from the father and all three
children did express it. The expression of glyceraldehyde-3-phosphate
dehydrogenase mRNA served as an internal standard for the amount
of RNA in each lane.
Immunohistochemical Evaluation of Bone Morphogenetic Protein
4 in Lymphoblastoid Cell Lines
Immunohistochemical analysis of cultured cells was used to determine
whether bone morphogenetic protein 4 was present in lymphoblastoid
cell lines from patients with fibrodysplasia ossificans progressiva
and normal subjects. The antibody we used recognizes bone morphogenetic
proteins 2 and 4, but because bone morphogenetic protein 2 mRNA
was not detected in these cells, antibody binding indicated the
presence of bone morphogenetic protein 4. This protein was detected
in the lymphoblastoid cells from 10 patients with fibrodysplasia
ossificans progressiva but not in 10 normal subjects (Fig. 5).
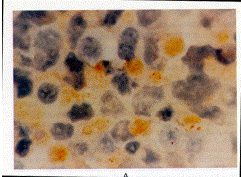
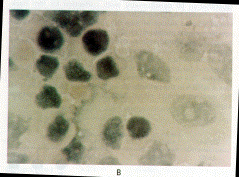
Figure 5. Expression
of Bone Morphogenetic Protein in Cells from a Patient with Fibrodysplasia
Ossificans Progressiva (Panel A) and a Normal Subject (Panel B)
(X1250).
The cells were stained with an antibody that detects bone morphogenetic
proteins 2 and 4. A positive immunoperoxidase reaction product
(brown staining) is seen only in the cytoplasm of lymphoblastoid
cells from the patient. Cell nuclei stain blue with the counterstain
(Wright's stain).
DISCUSSION
The bone morphogenetic proteins, a family of potent osteogenic agents in the TGF-ß superfamily of peptides, induce endochondral osteogenesis and fracture healing.13,15,19-28 These substances act by diffusion in a concentration-dependent manner to specify the fate of cells in embryogenesis and bone regeneration.15,27-30 Bone morphogenetic proteins are unique in their ability to induce the complete cellular program of endochondral osteogenesis at heterotopic sites in vivo.13,15,31,32 Our study provides evidence that overexpression of a bone morphogenetic protein is associated with a disabling disorder of osteogenesis in humans.
We previously proposed that overexpression of a bone morphogenetic protein gene may be involved in the gain of function leading to heterotopic ossification in patients with fibrodysplasia ossificans progressiva.10 The results of this study suggest a mechanism to explain the pathophysiology of heterotopic bone formation in this disorder. Since the half-life of bone morphogenetic protein is extremely brief (only a few minutes), it is unlikely that osteogenesis-inducing concentrations of bone morphogenetic protein 4 could be achieved at sites of osteogenesis unless the morphogen is delivered to those sites by circulating cells or manufactured at those sites by mesenchymal cells. As evidence, Chinese-hamster-ovary cells tranfected with bone morphogenetic protein 4 and implanted at soft-tissue sites in mice induce ectopic ossification at the sites of implantation but not at remote locations.31,32
We propose that lymphocytes capable of expressing bone morphogenetic protein 4 circulate in the peripheral blood of patients with fibrodysplasia ossificans progressiva and are recruited to connective tissue after soft-tissue injury.33,34 Type IV collagen, a primary constituent of the basement membrane of endothelial cells and muscle cells, avidly binds bone morphogenetic protein 4,35 resulting in increased local concentrations. At high concentrations, bone morphogenetic protein 4 acts as a morphogen13,15,19,36 capable of up-regulating its own mesenchymal expression22 and leading to the development of pre-osseous fibroproliferative lesions.13
The appearance of lymphocytes in the perivascular space of the earliest detectable lesion of fibrodysplasia ossificans progressiva6 provides support to the view that lymphocytes and perivascular cells are involved in the induction of osteogenesis.37,38
The stringent temporal and spatial patterns of postnatal heterotopic ossification in patients with fibrodysplasia ossificans progressiva are reminiscent of the patterns of mesenchymal-cell condensation during skeletal embryogenesis and suggest a common molecular basis for prenatal and postnatal osteogenesis.1,4,5,15 In humans postnatal osteogenesis occurs most commonly during healing of fractures. The creation of callus at a fracture site and heterotopic bone formation in fibrodysplasia ossificans progressiva follow nearly identical endochondral pathways7 involving lymphocytes6,37 and increases in the level of bone morphogenetic protein 4. 25,26
Dysregulation of bone morphogenetic protein during skeletal embryogenesis provides a tantalizing model for skeletal malformations in neonates with fibrodysplasia ossificans progressiva.5 In mice, naturally occurring mutations of bone morphogenetic protein genes result in congenital abnormalities of the skeleton and in postnatal abnormalities in fracture repair.15,23,24 Homozygous deletions of bone morphogenetic protein 5 result in dysmorphology of the axial skeleton and in abnormal fracture repair in short ear mice.23 Homozygous mutations of growth differentiation factor 5 result in dysmorphology of the appendicular skeleton in brachypodism mice.24 Fibrodysplasia ossificans progressiva in humans appears to be the only known example of a naturally occurring genetic disorder of osteogenesis that is associated with overexpression of a bone morphogenetic protein.
Our model raises many questions about lymphocytes, inflammation, tissue repair, and osteogenesis. What is the role of lymphocytes in osteogenic induction, bone development, fracture repair, and nongenetic forms of heterotopic osteogenesis? Do lymphocytes normally produce bone morphogenetic proteins? Is the overexpression of bone morphogenetic protein 4 a primary or secondary abnormality? Do the lymphocytes of patients with fibrodysplasia ossificans progressiva transport bone morphogenetic protein 4 to the site of osteogenic induction, or is the expression of bone morphogenetic protein 4 up-regulated locally at the site of osteogenesis?
The cause of lymphocytic activation in patients with fibrodysplasia ossificans progressiva is not known. Lymphoblastoid cell lines immortalized with Epstein-Barr virus can induce the transcription of genes that are not normally expressed in lymphocytes in vivo.39 Therefore, induction of the expression of bone morphogenetic protein 4 in lymphoblastoid cell lines from patients with fibrodysplasia ossificans progressiva could be the result of transformation by the Epstein-Barr virus or other cell-culture conditions. However, bone morphogenetic protein 4 was not overexpressed in identically treated lymphoblastoid cell lines from normal subjects, indicating a clear genetic difference between cells affected by fibrodysplasia ossificans progressiva and normal cells. The possibility of lymphocyte-mediated induction of heterotopic osteogenesis has broad implications for the study of hematopoiesis and skeletal biology.
Supported by grants from the International Fibrodysplasia Ossificans Progressiva Association, the American Heart Association, the Orthopaedic Research and Education Foundation (Johnson and Johnson research award), and the National Institutes of Health (R01-AR-41916).
REFERENCES
1. Cohen RIB, Hahn GV, Tabas JA, et al. The natural history of heterotopic ossification in patients who have fibrodysplasia ossificans progressiva: a study of forty-four patients. J Bone Joint Surg Am 1993; 75:215-9.
2. Connor JM, Skirton H. Lunt PW. A thrce generation family with fibrodpplasia ossificans progressive J Mod Genet 1993;30:687-9.
3. Kaplan FS, McCluskey W. Hahn G. Tabas JA, Muenke M, ZasloffMA. Genetic transmission of fibrodysplasia ossificans progressiva: report of a punily. J Bone Joint Surg Am 1993,75:1214-20.
4, Locke DM, ZasloffM, Peeper J. Cohen RIB, Kaplan FS. Age- and jointspecific risk of initial heterotopic ossification in patients who have fibrodysplasia ossificans progressive Clin Orthop 1994;301:243-8.
5. Connor JM, Evans DA. Fibrodysplasia ossificans progressiva: the clinical features and natural history of 34 patients. J Bone Joint Surg Br 1982;64:
6. Kaplan FS, Gannon FH, Shafritz AB, ZasloffMA, Shore EM. Acute lymphocytic infiltration in a very early lesion of fibrodysplasia ossificans progressive Calcif Tissue Int (in press). abstract.
7. Kaplan FS, Tabas PA, Gannon FH, Finkel G. Hahn GV, ZasloffMA. The hismpathology of fibrodysplasia ossificans progressiva: an endochondral process. J Bone Joint Surg Am 1993;75:220-30.
8. Lanchoney TF, Cohen RIB, Rocke DM, Zasloff MA, Kaplan FS. Permallcnt heterotopic ossification at the injection site after diphtheria-tetanus-pertussis immunizations in children who have fibrodysplasia ossificans progressiva. J Pediatr 1995,126:762-4.
9. Shah PB, Zasloff MA Drummond D, Kaplan FS. Spinal deformity in patients who have fibrodysplasia ossificans progressive J Bone Joint Surg Am 1994;76:1442-50.
10. Kaplan FS, Tabas JA, ZasloffMA. Fibrodpplasia ossificans progressiva: a clue from the fly? Calcif Tissue Int 1990;47:117-25.
11. Janoff HB, Tabas JA, Shore EM, et al. Mild expression of fibrodpplasia ossificans progressiva: a report of 3 cases. J Rheumatol 1995;22: 976-8.
12. Watt JL, Stephen GS. Lymphocyte culture for chromosome analysis. In: Roonq DE, Czepulkowski BH, ads. Human cytogenetics: a practical approach. Oxford, England: ILL Press, 1986:39-55.
13. Woznq JM, Rosen V, Celeste AS, et al. Novel regulators of bone formation: molecular clones and activities. Science 1988-242:1528-34.
14. Chomczynsh g Sacchi N. Single-step method of RNA isolation by add guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 1987;162:156-9.
15. Kingsley DM. The TGF-beta superfamily: new members, new receptors, and new genetic tests of function in different organisms. Genes Dev 199418:133-46.
16. Wang ZQ, Grigoriadis AE, Mohle-Steinlein U. Wagner ER A novel target cell for c-fos-induced oncogenesis: development of chondrogenic tumours in embryonic stem cell chimeras. EMBO J 1991;10:2437-50.
17. Jabs EW, Muller U. Li X, et al. A mutation in the homeodomain of the human MSX2 gene in a family affected with autosomal dominant craniosynostosis. Cell 1993,75:443-50.
18. Sambrook J. Fntsch EF, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press, 1989.
19. Urist MA Bone: formation by autoinduction. Science 1965,150:893-9
20. Jones CM Lyons KM, Hogan BL. Involvement of bone morphogeneuc protein-i (BMP-4) and Vgr-1 in morphogenesis and neurogenesis in the mouse. Development 1991;111:531-42.
21. Francis PH, Richardson MK, Bricked PM, Tickle C. Bone morphogenetic proteins and a signalling pathway that controls patterning in the developing chick limb. Development 1994,120:209-18.
22. Vainio S. Karavanova I, Jowett A, Thesleff I. Identification of BMP-4 as a signal mediating secondary induction between epithelial and mesenchymal tissues during early tooth development. Cell 1993;75:45-58.
23. Kingsley DM, Bland AE, Grubber JM, et al. The mouse short ear skeletal morphogenesis locus is associated with defects in a bone morphogenetic member of the TGF beta superfamily. Cell 1992-71:399-410.
24. Storm EE, Huynh TV, Copeland NG, Jenkins NA, Kingsley DM, Lee SJ. Limb alterations in brachypodism mice due to mutations in a new member of the TGF beta-superfamily. Nature 1994;368:639-43.
25. Nakase T. Nomura S. Yoshikawa H. et al. Transient and localized expression of bone morphogenetic protein 4 messenger RNA dunng fracture healing. J Bone Miner Res 1994,9:651-9.
26. Bostrom ME, Lane JM, Berberian WS, et al. Immunolocalization and expression of bone morphogenetic proteins 2 and 4 in fracture healing. J Orthop Res 1995;13:357-6Z
27. Johansson BM, Wales MV. Evidence for involvement of activin A and bone morphogenetic protein 4 in mammalian mesoderm and hematopoietic development. Mol Cell Biol 1995,15:141-51.
28. Wmnier G. Blessing M, Labosky PA, Hogan BL. Bone morphogenetic protein-4 is requited for mesoderm formation and patterning in the mouse. Genes Dev 1995;9:2105-16.
29. Padgett RW, Woznq JM, Gelbart WM. Human BMP sequences can confer normal dorsal-ventral patterning in the Drosophila embryo. Proc Natl Acad Sci U S A 1993,90:2905-9.
30. Zecca M, Basler K, Strum G. Sequential organizing activities of engrailed, hedgehog and decapentaplegic in the Drosophila wing. Development 1995;121:2265-78.
31. Takaoka K, Yoshikawa H. Hashimoto J. Ono K, Matsui M, Nakazato H. Transfilter bone induction by Chinese hamster ovary (CHO) cells transfected by DNA encoding bone morphogenetic protein-4. Clin Orthop 1994-300:269-73.
32. Shimizu K, Yoshikawa H. Matsui M, Masuhara K, Tahoh K. Periosteal and intratumorous bone formation in athymic nude mice by Chinese hamster ovary tumors expressing murine bone morphogenetic protein-4. Clin Orthop 1994;300:274-80.
33. Springer TA. Traffc signals for lymphocyte recirculauon and leukocyte emigration: the multistep paradigm. Cell 1994;76:301-14.
34. Buring K On the ongin of cells in heterotopic bone formation. Clin Orthop 1975; 110: 293-307
35. Reddi AH, Cunningham NS. Initiation and promotion of bone differentiation by bone morphogenetic proteins. J Bone Miner Res 1993,8: Suppl 2:S499-S502.
36. Katagiri T. Yamaguchi A, Komaki M, et al. Bone morphogenetic protein-2 converts the differentiation pathway of C2C12 myoblasts into the osteoblast lineage. J Cell Biol 1994;127:1755-66. [Erratum, J Cell Biol 1995;128:713.]
37. Einhorn TA, Majeska RJ, Rush EB, Levine PM, Horowitz MC. The expression of cytokine activity by fracture callus. J Bone Miner Res 199510:1272-81.
38. Brighton CT, Lorich DG, Kupcha R. Reilly TM, Jones AR, Woodbury RA II. The pericyte as a possible osteoblast progenitor cell. Clin Orthop 1992;275 :287-99.
39. Liebowitz D, Kieff E. Epstein-Barr virus. In: Roizman B. Whitley RJ Lopez C, eds. The human herpesviruses. New York: Raven Press, 1993: 107-72.
We are indebted to the following for gifts of cDNA clones Dr. Darwin Proctop (Thomas Jefferson Medical College, Philadelphia) for collagens type I and II, Dr. Thomas Kadesch (University of Pennsylvania, Philadelphia) for alkaline phosphatase, Dr. Se-Jin Lee (Johns Hopkins University, Baltimore) for growth-differennation factor 5, Dr. Dwight Bowler (Washington University, St Louis) for MSX-2, and Dry. John Wozney and Vicki Rosen (Genetics Institute, Cambridge, Mass) for generously providing bone morphogenetic protein and osteocalcin probes and antibody against bone morphogenetic proteins 2 and 4; to Heather Mitchell for establishing lymphoblastoid cell lines; to Dry. Leslie Sutton and Ann-Christine Duhaime for providing surgical specimens to establish primary cell lines; to Dr. Leslie Crofford for providing an established line of fibrodysplasia ossificans progressiva lesional cells; to Michael Haraschak for assistance with tissue culture; to Drs. Elizabeth Rand and Keyong Du for advice on laboratory techniques; to Dr. David Rocks for statistical analysis; to Gretchen Worden and the Mutter Museum of the College of Physicians of Philadelphia for archival photographs; to Joanne Deithorn for clinical photography to Frank Yue for assistance in preparing figures; to Drs. J. Michael Connor, William Gelbart, Victor McKusick and Marshall Urist for thoughtful and scholarly discussions during the course of this work and to Jeannie Peeper (president of the International Fibrodysylasia Ossificans Progressiva Association) and all the patients affected with fibrodysplasia ossificans progressiva for their steadfast courage in guiding us on this journey.
Return To Molecular Biology Main Page
![]()
©
Copyright 2000 Department of Biology, Davidson College, Davidson,
NC 28036
Send comments, questions, and suggestions to: macampbell@davidson.edu