Proc. Natl. Acad. Sci. USA
Vol. 88, pp. 9578-9582, November 1991
Biochemistry
(yeast / GAL4 / transcriptional activation / transcriptional silencing / dimerization)
CHENG-TING CHIEN*, PAUL L. BARTELÝ, ROLF STERNGLANZ*, AND STANLEY FlELDSݧ
Departments of *Biochemistry and Cell Biology and
ÝMicrobiology,
State University of New York, Stony Brook, NY 11794
§To whom reprint requests should
be addressed.
Communicated by Keith R. Yamamoto, August 2, 1991 (received for review May 14, 1991)
ABSTRACT We describe a method that detects proteins capable of interacting with a known protein and that results in the immediate availability of the cloned genes for these interacting proteins. Plasmids are constructed to encode two hybrid proteins. One hybrid consists of the DNA-binding domain of the yeast transcriptional activator protein GAL4 fused to the known protein; the other hybrid consists of the GAL4 activation domain fused to protein sequences encoded by a library of yeast genomic DNA fragments. Interaction between the known protein and a protein encoded by one of the library plasmids leads to transcriptional activation of a reporter gene containing a binding site for GAL4. We used this method with the yeast SIR4 protein, which is involved in the transcriptional repression of yeast mating type information. (i) We used the two-hybrid system to demonstrate that SIR4 can form homodimers. (ii) A small domain consisting of the C terminus of SIR4 was shown to be sufficient to mediate this interaction. (iii) We screened a library to detect hybrid proteins that could interact with the SIR4 C-terminal domain and identified SIR4 from this library. This approach could be readily extended to mammalian proteins by the construction of appropriate cDNA libraries in the activation domain plasmid.
Specific interactions between proteins form the basis of many essential biological processes. Additionally, transforming proteins of tumor viruses in many cases exert their effect through their interactions with cellular proteins; for example, the simian virus 40 (SV40) large tumor (T) antigen binds to the cellular proteins p53 and Rb (1, 2). Consequently, considerable effort has been made to identify those proteins that bind to proteins of interest. Typically, these interactions have been detected by using coimmunoprecipitation experiments in which antibody to a known protein is used to precipitate associated proteins as well. Such biochemical methods, however, result only in the identification of the apparent molecular mass of the associated proteins; obtaining cloned genes for these proteins is often a difficult process. In one approach, this problem has been circumvented by the use of purified proteins as probes against bacterial expression libraries, where a positive signal for an interacting protein is accompanied by the availability of the corresponding gene (3).
We have described a method by which a protein-protein
interaction is identified in vivo through reconstitution of the activity
of a transcriptional activator (4). The method is based on the properties
of the yeast GAL4 protein, which consists of separable domains responsible
for DNA-binding and transcriptional activation (5). Plasmids encoding two
hybrid proteins, one consisting of the GAL4 DNA-binding domain fused to
protein X and the other consisting of the GAL4 activation domain fused to
protein Y, are constructed and introduced into yeast. Interaction between
proteins X and Y leads to the transcriptional activation of a reporter gene
containing a binding site for GAL4.
In this paper, we demonstrate the utility of this in vivo approach
(designated the two-hybrid system) to screen a random library for an interacting
protein. Our test case is the yeast SIR4 gene product, a protein involved
in the transcriptional repression of the silent copies of mating type information
(6). We first showed by construction of GAL4 hybrids with SIR4 domains that
SIR4 is capable of dimer formation and that this dimerization can be mediated
by a small C-terminal fragment of SIR4. We then transformed yeast with both
a plasmid encoding the GAL4 DNA-binding domain fused to part of SIR4 and
a library of plasmids containing yeast genomic fragments fused to the GAL4
activation domain. By screening for transcription of a GAL4-dependent reporter
gene, we identified a plasmid from the library carrying a fragment of the
SIR4 gene. This method of identifying interacting proteins has obvious applications
in other systems.
MATERIALS AND METHODS
Yeast Strains and Methods. Yeast strains used were GGY1::171(7) and W303 (8). Yeast were grown in YEPD or selective minimal medium (9). Transformation was by the high-efficiency method of Schiestl and Gietz (10). ß-Galactosidase activity was assayed either on plates or in liquid; yeast transformants were replica-plated to SSX plates or liquid cultures were assayed for 4-nitrophenyl ß-D-galactoside cleavage as described (4). SSX plates contained 6.7 g of yeast nitrogen base, 14 g of agar, 40 mg of 5-bromo4-chloro-3-indolyl ß-D-galactoside, 100 ml of 1 M potassium phosphate (pH 7.0), 100 ml of 20% (wt/vol) sucrose, 100 ml of 10x amino acids minus leucine and histidine (9), and H2O to 1 liter. Escherichia coli MH4 (11) contained the leuB mutation, which can be complemented by the yeast LEU2 gene.
Construction of GAL4 Activation Domain Plasmids. Plasmid pGX13 was derived from 401-N (12) and pKLC15 (13) and contains as a HindIII fragment sequences encoding the first five codons from SV40 T antigen, the nuclear localization signal from T antigen, amino acids 11-34 from T7 RNA polymerase, and amino acids 768-881, containing an activation domain, from GAL4. Site-directed mutagenesis was used to destroy the termination codon of GAL4 and create a Bgl II site at this location. The HindIII fragment was inserted into the unique HindIII site of pAE7, which is pAAH5 (14) that has had its two BamHI sites removed by sequentially linearizing with BamHI and filling-in the overhang with DNA polymerase I (Klenow fragment). Finally, oligonucleotides containing the BamHI sites in all three frames were inserted into the Bgl II site to generate the three pGAD vectors. Our initial activation domain plasmids (pGAD1, pGAD2, and pGAD3) have the HindIII fragment from pGX13 inserted into pAE7 in the orientation such that transcription of the fusion protein is driven by a promoter lying within sequences for termination of ADH1 transcription. Plasmids have also been constructed in the opposite orientation (pGADlF, pGAD2F, and pGAD3F) in which transcription of the fusion protein gene is driven by the ADH1 promoter. Whereas the ADDS promoter results in additional expression of the fusion protein, it leads to only minor changes in the level of GAL1-lacZ transcription derived by interaction of the two hybrids.
Construction of GAL4 Protein Fusions. Plasmid pES15 (from Elisa Stone, State University of New York at Stony Brook) containing the entire SIR1 sequence in pUC18 was cut at the Nru I site 52 base pairs 5' to the initiator ATG and an oligonucleotide containing an Xho I site was inserted. The resultant plasmid was cut with Xho I and the SIR1 fragment was ligated to Sal I-digested pMA424 (15) to create pKL5. An Xho I fragment containing the entire SIR2 sequence was prepared by the polymerase chain reaction from pJM100 (16) and was ligated to Sal I-digested pMA424 to produce pCTC7. pJR104 (17), which carries the SIR3 gene, was digested with Sal I and the SIR3-containing fragment was ligated into pUC18. The resulting plasmid (pKL3) was digested first with Bcl I and HindIII and then with Xho II and HindIII and the two appropriate fragments were ligated into BamHI-digested pGAD3 to yield pKL13. The SIR4 gene was subcloned as a Hind III fragment from pJR643 (from Jasper Rine, Univ. of California, Berkeley) into pUC18 to produce pCTC15. A BamH I fragment encoding amino acids 839-1358 was inserted into the BamHI site of pMA424 to generate pCTC17. This same BamHI fragment was used to generate the activation domain hybrid pCTC18. The SIR4 fragment corresponding to amino acids 1262-1358 was generated as a BamHI fragment by the polymerase chain reaction using as template pJR643. This fragment was inserted into the BamHI site of pMA424 to generate pCTC23 and into pGAD2 to generate pCTC24. The SNF4 gene was derived from plasmid pSL321 (18). The Cla I site at codon 20 and the polylinker Kpn I site after codon 321 (18) were converted to BamHI sites by insertion of oligonucleotide adaptors, and the resultant BamH I fragment was inserted into pGAD3 to generate pCTC14.
Construction of Yeast Genomic Libraries. Yeast genomic DNA was isolated from strain W303 by spheroplasting the cells with Zymolyase and adding 2 vol of lysis buffer [3% wt/vol) Sarkosyl/0.5 M Tris Cl/0.2 M EDTA, pH 9] to liberate the cell contents. Cell debris was pelleted after addition of 0.5 vol of 5 M potassium acetate, followed by ethanol precipitation of the DNA. The DNA was partially digested with Sau3A and was fractionated on 5-30% (wt/vol) sucrose gradients to isolate fragments from 2 to 6 kilobases in size. This DNA was ligated to pGAD1, pGAD2, or pGAD3 DNA that had been digested with BamHI and dephosphorylated with calf intestinal alkaline phosphatase. Ligation mixes are used to transform E. coli DH5 by electroporation. After growth on ampicillin-containing plates overnight, colonies were pooled and frozen at -70°C. These frozen stocks were used to grow the ampicillin-resistant transformants for preparation of plasmid DNA.
Screening Activation Domain Libraries. Yeast strain GGY1::171, which is His- and Leu-, was transformed simultaneously with both a DNA-binding domain plasmid and one of the libraries of genomic DNA fragments. After 3-4 days growth on SD-His-Leu plates (9), His+, Leu+ transformants were replica-plated to SSX plates and were incubated until blue colonies appeared. False positives due to cloning of the GAL4 gene into the pGAD vectors were eliminated by one of several strategies. In one approach, blue colonies were grown to saturation nonselectively in YEPD liquid and replated on YEPD plates. Replica plating of these plates to SD-His and SD-Leu identified colonies that had lost one or both plasmids, and replica plating to SSX plates (supplemented with histidine and leucine) indicated whether ß-galactosidase was expressed. Any colonies that remained blue after loss of the DNA-binding domain plasmid were eliminated. In a second approach, the blue colonies were grown in SD-Leu liquid medium and plasmid DNA was isolated from the culture (19) and transformed into E. coli by electroporation. The activation domain (pGAD) plasmids from the library were identified by their ability to complement an E. coli leuB mutation due to the plasmid-borne LEU2 gene. These plasmids were then individually reintroduced into yeast GGY1::171 either alone or with the DNA-binding domain plasmid. Yeast transformants that expressed ß-galactosidase when they contained the library plasmid alone were eliminated. Alternatively, the activation domain plasmid was cut with Xho I and Sal I to assay for a 1.0-kilobase fragment diagnostic of the GAL4 gene (20). Plasmids encoding interacting proteins were sequenced by the dideoxynucleotide method (21) using a primer corresponding to codons 758-763 of the GAL4 sequence.
RESULTS
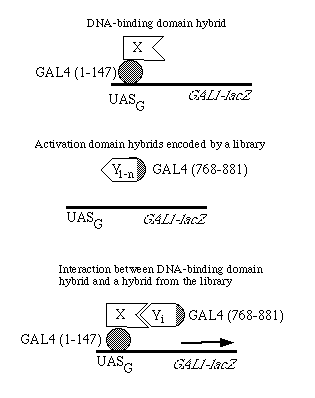
FIG. 1. Strategy to detect interacting proteins using the two hybrid system. UASG is the upstream activation sequence for the yeast GAL genes, which binds the GAL4 protein. The libraries of activation domain hybrids are constructed in the pGAD vectors.
Construction of Activation Domain Vectors. The principle of using two hybrid proteins to detect protein-protein interactions is shown in Fig. 1. A gene fusion is first generated that encodes the protein under study (protein X) as a hybrid with the DNA-binding domain of GAL4. This domain enables the hybrid to localize (22) to the nucleus and bind in a site-specific fashion to DNA (5). For the method to work, the protein X domain of this hybrid must fail to activate transcription of the reporter gene. Second, a gene fusion is introduced that encodes a protein Y sequence as a hybrid with the GAL4 activation domain; this hybrid also enters the nucleus. Protein Y may be encoded by a known gene or represent a library of sequences (Y1, Y2, Y3, . . ., Yn). Any activation domain hybrid carrying a protein Y sequence that can bind to protein X might be capable of reconstituting proximity of the two GAL4 domains in a manner that activates transcription of a reporter gene.
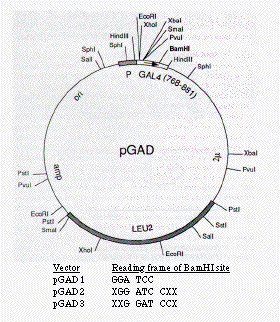
FIG. 2. Restriction map of the activation domain plasmid. The GAL4 activation
hybrid is transcribed from a promoter labeled P and contains in addition
a nuclear localization signal from SV40 T antigen, which is not indicated.
The three vectors differ in the reading frame of the BamHI cloning site,
as indicated. amp, Ampicillin resistance gene; ori, origin of replication
from pBR322.
In the previous report from this laboratory (4) using the two-hybrid system to detect the interaction of the yeast SNF1 and SNF4 proteins, the activation domain hybrid consisted of the yeast SNF4 protein at its N terminus and the GAL4 activation domain at its C terminus. Although the C-terminal location for the GAL4 activation domain corresponds to its normal position within the GAL4 protein, this location is inconvenient for the construction of a library of activation domain hybrids. We therefore determined whether the GAL4 activation domain (amino acids 768-881) could function at the N terminus of a protein fusion. This placement allows the use of the same vector-encoded yeast promoter and initiation codon for each gene fusion and requires a single fusion joint between the activation domain and a heterologous protein. In addition, we included within the activation domain fusion a sequence for nuclear targeting. We constructed a set of three plasmids, pGAD1, pGAD2 and pGAD3, as shown in Fig. 2, that differ only in the reading frame of the unique BamHI site. These vectors include an initiation codon, the nuclear localization signal from SV40 T antigen (23), which has been shown to function in yeast (12, 24), and the GAL4 activation domain (residues 768-881) followed by the BamHI cloning site.

Values for ß-galactosidase activity (28) are the mean of assays on at least two transformants, each assayed at least twice. The standard errors were typically 30% of the mean for values >2 units.
We used a pGAD vector to construct a hybrid between the GAL4 activation domain and the yeast SNF4 protein to test for the interaction between the SNF1 and SNF4 proteins. As shown in Table 1, in cells carrying the SNF1 protein in the DNA-binding domain plasmid, the SNF4 hybrid in plasmid pGAD3 led to a substantial level of GAL1-LacZ activity. This activity was comparable to that observed with the original SNF4 hybrid, which carried the GAL4 activation domain at its C terminus (4). This reconstruction experiment indicated that the pGAD vectors were suitable for testing other defined protein combinations and as recipients in the construction of libraries.
SIR Protein Interactions. The mating type of Saccharomyces cerevisiae is determined by the genetic information present at the MAT locus. Two other copies of mating type information, HML and HMR, are kept transcriptionally silent and serve as donors for transposition of sequences to MAT. This transcriptional repression is due to the specific action of four SIR gene products (SIR1 through SIR4) and the involvement of the proteins ABF1, RAP1, and histone H4, which have additional roles in the yeast cell (for review, see ref. 25). The mechanism of silencing and the biochemical interactions among these proteins are unknown.

ß-Galactosidase activity was determined as in Table 1.
To determine whether SIR proteins can bind to each other as indicated by a transcriptional signal in the two-hybrid system, we constructed a number of hybrid genes encoding SIR proteins fused to one of the GAL4 domains and introduced these genes singly or in pairwise combinations into a yeast reporter strain (Table 2). When present as fusions with the GAL4 DNA-binding domain, SIR1 (line 1), SIR2 (line 4) and SIR4 (lines 7 and 8) failed to activate transcription of GAL1-lacZ, indicating that these regions of the SIR proteins do not function as activation domains. We did not detect a signal for interaction of SIR1 with SIR3 or SIR4 (lines 2 and 3), SIR2 with SIR3 or SIR4 (lines 5 and 6), or SIR4 with SIR3 (line 9). Only with SIR4 present in both the DNA-binding domain and activation domain plasmids (line 10) was GAL1-lacZ transcription significantly above background. We note that we constructed only five of the eight possible GAL4 fusions with SIR proteins and assayed only 6 of 16 possible combinations. Lack of a signal is not strong evidence against a specific interaction; the hybrid proteins might not be stable, they might not include the residues necessary for interaction or the GAL4 domains might occlude a site of interaction. For SIR1 and SIR2, however, the hybrids with the GAL4 DNAbinding domain are capable of complementing sir1 and sir2 mutations, respectively, indicating that the protein fusions are sufficiently stable and active to cooperate with the other proteins required for transcriptional silencing. For SIR4, the hybrids contain no more than 520 residues of this 1358-residue protein and do not complement a sir4 mutation. However, the SIR4 hybrids exhibit an activity designated anti-SIR (26), in which overexpression of the C-terminal end of the SIR4 protein causes a dominant derepression phenotype.
The SIR4 constructions that resulted in a transcriptional signal from the reporter gene both contained the C-terminal 520 amino acids of SIR4. It has been noted that the most C-terminal region of SIR4 contains 12 heptad repeats and a similarity to human nuclear lamins (27). We therefore tested whether fragments of SIR4 consisting of essentially only these repeats (the C-terminal 97 amino acids of SIR4) were capable of a transcriptional signal indicating interaction (Table 2, line 11). The similar level of GAL1-lacZ transcription we detect with these small hybrids suggests that one SIR4 protein contacts another through a coiled-coiled interaction mediated by the heptad repeats.
Screening of an Activation Domain Library. Based on the demonstration of the SIR4-SIR4 interaction in the two hybrid system, we sought to determine whether this or other interactions could be detected by screening a library of total sequences present in the activation domain plasmid. With SIR4 as a fusion with the GAL4 DNA-binding domain, any protein encoded by an activation domain fusion that can interact with SIR4 might reconstitute GAL4 activity. We note that the library must be constructed in the activation domain plasmid to avoid detecting random (and abundant) sequences that can activate transcription when fused to a DNA-binding domain (15).
Each pGAD vector was ligated to a size-fractionated partial Sau3A digest of yeast genomic DNA, generating 2 x 106 individual transfornlants in E. coli. Colonies containing library constructions in each pGAD vector were pooled separately, and plasmid DNA was prepared. Yeast were cotransformed with a mixture of DNAs containing equal amounts of GAL4-(1-147)-SIR4-(839-1358) and one of the pGAD Sau3A libraries, selecting for both histidine and leucine prototrophy. This protocol was preferred over sequential transformation of first the DNA-binding domain plasmid and then the library because we have observed some instability of certain plasmids encoding DNA-binding domain hybrids. The cotransformation protocol minimizes the amount of time transformants are incubated before they are assayed for ß-galactosidase activity. Transformants were replica-plated to medium containing 2% sucrose, amino acid supplements lacking histidine and leucine, and 5-bromo-4-chloro-3-indolyl ß-D-galactoside (40 µg/ml). Approximately 1 transformant per 14,000 turned blue by 5 days, indicating transcription of GAL1-lacZ. In addition, we note that the activation domain library transformed alone caused a background of approximately the same ratio of blue colonies, which results from cloning of the GAL4 gene in the pGAD vectors. (The high background is due to the fact that the GAL4 gene can be present anywhere within the insert sequence and need not be in a defined position, orientation, and reading frame as does the gene for an interacting protein.)
Fifteen positive transformants (of a total of 220,000 transformants) were categorized as to whether GAL1-lacZ transcription required the presence of both hybrids. In one protocol, cells were grown nonselectively and screened for a return to histidine or leucine auxotrophy, indicating loss of the marker on the DNA-binding or activation domain plasmid, respectively. A comparison of the histidine and leucine requirements with ß-galactosidase expression indicated whether both plasmids were required for GAL4 function. In a second approach, each of the library plasmids was isolated and reintroduced into yeast with or without the DNA-binding domain plasmid, and these transformants were tested for ß-galactosidase activity. Two of the 15 positives required both plasmids to reconstitute GAL4 function. DNA sequence analysis of the insert from one of these activation domain hybrids indicated that it encoded a portion of the SIR4 gene, beginning at a Sau3A site corresponding to amino acid residue 1205, and restriction digestion analysis indicated that the insert contained the rest of the SIR4 gene. This hybrid contains 154 amino acids of the SIR4 protein, slightly more than the small C-terminal SIR4 fragment that we had tested. ß-Galactosidase activity of this hybrid from the library (Table 2, line 12) showed a level comparable to that observed in the reconstruction experiments. The other plasmid that required the fusion of the GAL4 DNA-binding domain and SIR4 for GAL4 function carries the gene designated SFI1 (for SIR4-interacting protein), whose partial sequence does not correspond to any yeast gene sequence in available data bases (February 1991). The SFI1-containing plasmid alone was inactive for GAL1-lacZ transcription (Table 2, line 13) but with GAL4-(1-147)-SIR4-(839-1358) produced 96 units of activity (Table 2, line 14).
The yeast genome contains ~50,000 Sau3A fragments which can be ligated in either orientation to yield 100,000 possible fusion joints in each of the pGAD libraries. If a gene encoding an interacting protein contains a single appropriate Sau3A site, the probability of detecting the correct GAL4 hybrid in a given library is 90% by screening 230,000 transformants and 99% by screening 460,000 transformants. Thus it is possible that there are additional proteins capable of interacting with SIR4 that would be found with additional library screening.
DISCUSSION
The approach of using two GAL4 hybrids to detect protein-protein interactions has been extended to three additional applications. (i) It can be used with available genes to test pairwise combinations for interaction. Such testing of SIR proteins provides strong evidence for a SIR4-SIR4 complex. (ii) A positive signal for interaction allows a rapid means to identify the specific domains responsible for the proteinprotein contacts. For SIR4, hybrids carrying only 7% of the protein were used to demonstrate that the contacts appear to be mediated by a series of heptad repeats present at the C terminus. (iii) A library of total sequences fused to the activation domain can be screened to detect a plasmid encoding an interacting protein. We used such a yeast library to identify a SIR4 insert and another gene.
By testing a series of pairwise combinations of SIR proteins in the two-hybrid system, we detected a signal for interaction only between SIR4 and itself. This result suggests that SIR4 may function in silencing as a dimer or higher-order multimer. It also provides a possible explanation for the anti-SIR activity of SIR4 C-terminal fragments (26), observed as derepression of the silent mating type loci in a SIR+ strain: the overproduced SIR4 fragments may bind to the wild-type SIR4 protein and prevent it from exerting its activity.
In using the two-hybrid approach for screening a library of activation-domain hybrids, a major advantage is the immediate availability of the cloned gene for the interacting protein. In addition, only a single plasmid construction is required to use this method; there is no necessity to prepare either antibody or purified protein for the biochemical detection of interactions. The interactions are detected in vivo, under conditions that may be similar to those that occur naturally. Because the background of GAL1-lacZ transcription is negligible, even interactions that reconstitute only a low level of GAL4 function produce a detectable signal. Thus it may be possible to detect transient interactions, such as those occurring during only a limited portion of the cell cycle. Finally, the stability of the ß-galactosidase protein allows the accumulation of a weak signal over time.
Comparison of the signal generated by the combination of GAL4-(1-147)-SIR4-(839-1358) or GAL4-(1-147)-SIR4-(1262-1358) with three other SIR4 hybrids with the activation domain indicates that all six combinations are similarly effective at reconstituting GAL4 function (Table 2 and unpublished results). This result further supports the hypothesis that the structure of the two interacting hybrids can be highly variable, with the major requirement being only the presence of the interacting domains. Such flexibility suggests that numerous other proteins may be detected in this system. Reconstruction experiments indicate that such interacting protein pairs as p53-SV40 large T antigen (unpublished results), retinoblastoma protein-SV40 large T antigen (T. Durfee and W.-H. Lee, personal communication), and the ß and gamma subunits of the yeast pheromone-responsive guanine nucleotide binding protein (K. Clark and M. Whiteway, personal communication) give positive signals in this system. In addition, other screens for interacting proteins using the libraries described here have detected candidate yeast proteins capable of binding to p53 (unpublished results) and RAP1 (C. Hardy and D. Shore, personal communication). Although interaction in these experiments has been detected by screening for blue colony color, it should be feasible to use this method in a strain carrying a GAL4-dependent selectable gene. Such a strain would allow the assaying of cDNA libraries of high complexity while requiring the use of relatively few plates. This approach might thus lead to the identification of various interacting proteins of mammalian origin.
Acknowledgements: We thank Mark Swanson for some of the plasmids used in these experiments and Joe Lipsick for comments on the manuscript. This work was supported by U.S. Public Health Service Research Grants GM28220 to R.S. and CA54699 to S.F. and by grants from the Procter and Gamble Company and New York State Science and Technology Foundation to S.F.
Abbreviations: SV40, simian virus 40; T, tumor.
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked "advertisement" in accordance with 18 U.S.C. §1734 solely to indicate this fact.
References
1. Lane, D. P. & Crawford, L. V. (1979) Nature (London) 278, 261-263.
2. DeCaprio, J. A., Ludlow, J. W., Figge, J., Shew, J.-Y., Huans, C.-M., Lee, W.-H., Marsilio, E., Paucha, E. & Livingston, D. M. (1988) Cell 54, 275-283.
3. Nelbock, P., Dillon, P. J., Perkins, A. & Rosen, C. A. (1990) Science 248, 1650-1653.
4. Fields, S. & Song, O. (1989) Nature (London) 340, 245-246.
5. Keegan, L., Gill, G. & Ptashne, M. (1986) Science 231, 699-704.
6. Rine, J. & Herskowitz, I. (1987) Genetics 116, 9-22.
7. Gill, G. & Ptashne, M. (1987) Cell 51,121-126.
8. Thomas, B. J. & Rothstein, R. (1989) Cell 56, 619-630.
9. Sherman, F., Fink, G. R. & Hicks, J. B. (1986) Methods in Yeast Cenetics (Cold Spring Harbor Lab., Cold Spring Harbor, NY).
10. Schiestl, R. H. & Gietz, R. D. (1989) Curr. Genet. 16, 339-346.
11. Hall, M. N., Hereford, L. & Herskowitz, I. (1984) Cell 36,1057-1065.
12. Benton, B. M., Eng, W.-K., Dunn, J. J., Studier, F. W., Sternglanz, R. & Fisher, P. A. (1990) Mol. Cell. Biol. 10, 353-360.
13. Ma, J. & Ptashne, M. (1987) Cell 48, 847-853.
14. Ammerer, G. (1983) Methods Enzymol. 101, 192-201.
15. Ma, J. & Ptashne, M. (1987) Cell 51, 113-119.
16. Mullen,J. R.,KaynejP. S.,Moerschell,R. P.,Tsunasawa,S., Gribskov, M., Colavito-Shepanski, M., Grunstein, M., Sherman, F. & Sternglanz, R. (1989) EMBO J. 8, 2067-2075.
17. Kimmerly, W. J. & Rine, J. (1987) Mol. Cell. Biol. 7, 42254237.
18. Celenza, J. L., Eng, F. J. & Carlson, M. (1989) Mol. Cell. Biol. 9, 5045-5054.
19. Hoffman, C. S. & Winston, F. (1987) Gene 57, 267-272.
20. Laughon, A. & Gesteland, R. F. (1984) Mol. Cell. Biol. 4, 260-267.
21. Sanger, F., Nicklen, S. & Coulson, A. R. (1977) Proc. Natl. Acad. Sci. USA 74, 5463-5467.
22. Silver, P. A., Keegan, L. P. & Ptashne, M. (1984) Proc. Natl. Acad. Sci. USA 81, 5951-5955.
23. Kalderon, D., Roberts, B. L., Richardson, W. D. & Smith, A. E. (1984) Cell 39, 499-509.
24. Nelson, M. & Silver, P. (1989) Mol. Cell. Biol. 9, 384-389.
25. Alberts, B. M. & Sternglanz, R. (1990) Nature (London) 344, 193-194.
26. Marshall, M., Mahoney, D., Rose, A., Hicks, J. B. & Broach, J. R. (1987) Mol. Cell. Biol. 7, 4441-4452.
27. DiSey, J. F. X. & Stillman, B. (1989) Nature (London) 342, 24.
28. Miller, J. H. (1972) Experiments in Molecular Genetics (Cold Spring Harbor Lab., Cold Spring Harbor, NY).
Return To Molecular Biology Main Page
![]()
© Copyright
2000 Department of Biology, Davidson College, Davidson, NC 28036
Send comments, questions, and suggestions to: macampbell@davidson.edu