The Journal of Biological Chemistry Vol. 267, No. 13, Issue
of May 5, pp. 9321-9325, 1992
© 1992 by The American Society for Biochemistry and Molecular
Biology, Inc.
(Received for publication, December 23, 1991)
From the Departments of +Biology and §Medicine, The
Johns Hopkins University, Baltimore, Maryland 21218
¶ To whom correspondence should be sent. Tel.: 410-516-5174;
Fax: 410-516-5213.
The sarcoplasmic/endoplasmic reticulum slow-twitch or cardiac Ca2+-ATPase (SERCA2) is expressed as two forms (SERCA2a and SERCA2b) which vary at their extreme carboxyl termini. SERCA2a and SERCA2b are derived from alternatively spliced primary transcripts of the same gene. These two alternative carboxyl termini are highly conserved in mammals (Eggermont, J. A., Wuytack, F., De Jaegere, S., Nelles, L., and Casteels, R. (1980) Biochem. J. 260, 767-761; Lytton, J., and MacLennan, D. H. (1988) J. Biol. Chem. 263, 16024 - 16031) and birds (Campbell, A. M., Kessler, P. D., Sagara, Y., Inesi, G., and Fambrough, D. M. (1991) J. Biol. Chem. 266, 16060 - 16066). The topology of SERCA2a is believed to be identical to the fast-twitch Ca2+-ATPase (SERCA1) with 10 membrane-spanning domains. Based on hydropathy analysis, the extended carboxyl terminus of SERCA2b is predicted to span the endoplasmic reticulum (ER) membrane an additional (i.e. 11th) time. We have added the human c-myc epitope, a 10-amino acid sequence recognized by monoclonal antibody 9E10, onto the carboxyl termini of SERCA2a and SERCA2b to test whether or not their carboxyl termini are on the same side of the ER membrane. The added epitopes do not appear to disrupt topology as judged from unaltered Ca2+ transport. Immunocytochemical studies demonstrate that SERCA2a and SERCA2b have their carboxyl termini on opposite sides of the ER membrane; SERCA2a's is in the cytosol and SERCA2b's is in the ER lumen.
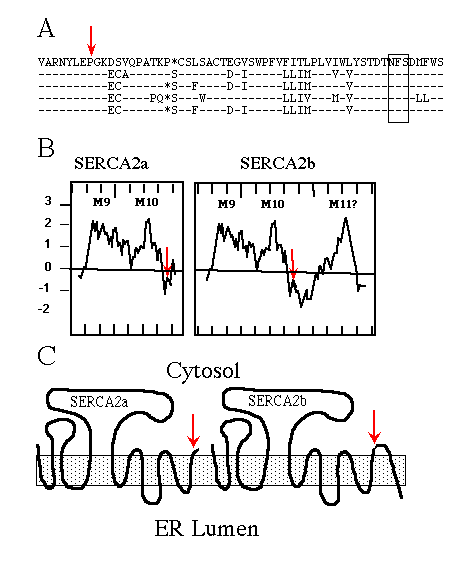
Immunological experiments support the topological model of SERCA1 having 10 membrane-spanning domains (Clarke et al., 1990; Matthews et al., 1990), originally proposed by MacLennan et al. (1985) . When SERCA2b was first sequenced (Lytton and MacLennan, 1988; Gunteski-Hamblin et al., 1988), hydropathy analysis suggested it had an additional (i.e. 11th) membrane-spanning domain. The same was found for the avian SERCA2b (Campbell et al., 1991). Fig. 1A shows the high percentage of amino acid sequence identity between chicken, rat, human, rabbit, and pig SERCA2b. Hydropathy plots (Fig. 1B; Kyte and Doolittle, 1982) of avian SERCA2a and SERCA2b, beginning with the last two common membrane-spanning domains, illustrate the point that SERCA2b might span the ER membrane once more than SERCA2a. The resulting topology is depicted in Fig. 1C.
FIG. 1. A, comparison of partial SERCA2b sequences using
the one-letter amino acid code, from top to bottom: chicken, rat, human,
rabbit, and pig. The dashes indicate identical residues; the asterisks are
gaps to maximize alignment; the red arrow indicates the alternative splice
site; and the box encloses the conserved asparagine-linked glycosylation
consensus sequence NFS.
B, hydropathy plots, using the Kyte and Doolittle (1982) program
(with a window set for 12 amino acids) of chicken SERCA2a and SERCA2b beginning
with amino acid 920. Positive numbers are hydrophobic, and negative numbers
are hydrophilic; hash marks on the X-axis demark units of 10 amino acids;
M9, M10, and M11 indicate the predicted membrane-spanning domains, the red
arrows indicate the alternative splice sites.
C, cartoons depicting SERCA2a and SERCA2b topologies if the
two forms have 10 and 11 membrane-spanning domains, respectively. The shaded
box represents the ER membrane, and the red arrows mark the alternative
splice site and thus sequence divergence.
To determine whether or not the carboxyl termini of SERCA2a and SERCA2b are on opposite sides of the ER membrane, we combined immunocytochemical techniques and a procedure to selectively permeabilize the plasma membrane with the bacterial toxin streptolysin-O (SLO).2 Initially, we employed polyclonal antisera specific for SERCA2a or SERCA2b termini (generously provided by F. Wuytack and R. Casteels (Katholiecke Universiteit, Leuven, Belgium); Eggermont et al., 1990; Wuytack et al., 1989) but were unable to get strong signals. Therefore, we decided to add onto the carboxyl termini of SERCA2a and SERCA2b a 10-amino acid sequence (EQKLISEEDL) derived from the human oncogene product c-myc , recognized by mAb 9E10 (Evan et al., 1985). These epitope-tagged constructs, SERCA2a-myc and SERCA2b-myc , were used to determine the location of the carboxyl termini.
EXPERIMENTAL PROCEDURES
c-myc Tag cDNA Constructs - The endogenous stop codons in the cDNAs encoding SERCA2a and SERCA2b were deleted and replaced by the c-myc tag encoding nucleotides. A polymerase chain reaction (PCR)3-based technique was used to add on the c-myc encoding nucleotides, and conditions were as recommended by the Amplitaq DNA polymerase instructions (Perkin-Elmer Cetus). Thirty temperature cycles of 95° C, 50° C (for 1 min), and 72° C (for 20 s) were performed to produce the c-myc -tagged SERCA2a and SERCA2b carboxyl termini encoding cDNA. The c-myc tags encoding PCR primers were as follows: SERCA2a-myc , 5' TGTGTGGGTACCTAGAGGTCTTCCTCAGAGATCAGCTTCTGCTC 3', SERCA2b-myc , 5' TGTGTGGGTACCTAGAGGTCTTCCTCAGAGATCAGCTTCTGCTCAGACCAGAACATATCAA AG 3'. These primers contain a KpnI site for cloning, a stop codon, the c-myc epitope (EQKLISEEDL) encoding nucleotides, and 20 bases of the appropriate avian sequence encoding the carboxyl terminus of either SERCA2a or SERCA2b. The other primer was derived from the cDNA sequence on the 5' side of the unique internal SalI site (Campbell et al., 1991). The PCR products were digested with KpnI and SalI, purified and cloned into pBluescript (Stratagene), and sequenced to ensure that there were no mutations. These were then cloned into the internal SalI site of SERCA2. The resulting SERCA2a-myc and SERCA2b-myc cDNAs were cloned into the KpnI site of the expression vector described below.
Expression in Tissue Culture - cDNAs of SERCA2a, SERCA2b, SERCA2a-myc , and SERCA2b-myc were cloned into the KpnI site of the expression vector pcDL-SRa296 (Takebe et al., 1988). COS-1 cells were transfected using DEAE dextran and 10% dimethyl sulfoxide as in Campbell et al. (1991).
Microsome Preparation - As in Campbell et al. (1991), transfected COS-1 cells were harvested and then homogenized with a Dounce homogenizer. The suspension was centrifuged at 10,000 x g for 20 min. and the supernatant was centrifuged at 100,000 x g for 60 min. The pellet was resuspended with 50 mM MOPS (pH 7.0), 10% sucrose and frozen in liquid nitrogen for later use. Total microsomal protein concentrations were determined with the BioRad protein assay reagent.
Immunoblot Analysis - 15 µg of microsomal protein from cells transfected with SERCA2a, SERCA2a-myc , SERCA2b, and SERCA2b-myc were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis on an 8% polyacrylamide gel (Laemmli, 1970), transferred to nitrocellulose, and incubated in 10 mM Tris (pH 7.4), 150 mM NaCl, 5% dry milk, 0.02% Tween 20 (Fisher), and 0.01% Antifoam A (Sigma), and either 5 µg/ml mAb 3H2, specific to avian SERCA2 (CaS-3H2- Kaprielian and Fambrough, 1987) or 10 µg/ml mAb 9E10, specific for the human c-myc epitope (Evan et al., 1985). The bound antibodies were detected as described in the enhanced chemiluminescence protocol (Amersham Corp.). Expression levels of all forms of SERCA2 were measured by 125I-labeled mAb 3H2 binding as in Campbell et al. (1991). Wild-type SERCA2a and SERCA2a-myc expression levels were comparable, as were wild-type SERCA2b and SERCA2b-myc . However, SERCA2a and SERCA2a-myc expression levels were about 60% of SERCA2b and SERCA2b-myc expression levels. This is consistent with previous results (Campbell et al., 1991).
Calcium Uptake Assay - 5.0 ml of the reaction mixture (20 mM MOPS, pH 7.0, 60 mM KCl, 5 mM MgCl2, 5 mM sodium oxalate, 0.2 mM EGTA, 0.2 mM CaCl2 (with 0.4 µCi/ml 45Ca2+) and 10 µg/ml microsomal protein) was equilibrated at 25° C for 5 min before 2.5 mM ATP was added at time zero. 1.0-ml aliquots were removed at timed intervals, filtered (0.45 µm pore size; Millipore), and washed with 12 ml of 10 mM MOPS (pH 7.0), 2 mM LaCl3. A liquid scintillation counter was used to determine amounts of 45Ca2+ on the filter. For thapsigargin (Bethesda Research Laboratories) inhibition, 20 nM thapsigargin was present during the 5-min incubation period. The difference between Fig. 3A and Fig. 3B in the nanomoles of Ca2+ transported per mg of total microsomal protein is probably a result of variations in transfection efficiency and/or protein accumulation.
Immunofluorescence - Cells were plated on multiple coverslips placed in the same 100-mm tissue culture dish to ensure the uniformity of transfections. Therefore, cells depicted in Fig. 4, a, c, and e (SERCA2a-myc ) were transfected simultaneously, as were the cells in Fig. 4, b, d, and e (SERCA2b-myc ). Transfected cells were permeabilized with SLO (Burroughs Welcome, Research Triangle Park, NC) by a method modified from Gravotta et al. (1990). In order to preserve the reticular morphology of the ER, the SLO was used at 1.6 units/ml in a sodium-free buffer (25 mM HEPES, pH 7.4, 2.5 mM magnesium acetate, 25 mM KCl, 250 mM sucrose). After the cells were permeabilized, they were fixed in buffered 1% formaldehyde for 10 min at room temperature and blocked with buffered 1% bovine serum albumin and 5 mg/ml L-lysine. Then the cells were incubated with mAb 9E10 or mAb 3H2 in buffered 1% BSA either with or without 0.25% saponin as indicated, followed by a fluorescein-conjugated goat anti-mouse secondary antibody, and photographed on an epifluorescence microscope. Cells labeled for immunofluorescence microscopy are referred to as "stained."
RESULTS AND DISCUSSION
To verify the cDNA constructs and Ca2+-ATPase identities, microsomal proteins derived from COS-1 cells transfected with cDNAs encoding SERCA2a, SERCA2a-myc , SERCA2b, or SERCA2b-myc were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to nitrocellulose, and probed with mAb 3H2 or mAb 9E10 (Fig. 2). mAb 3H2 identified the wild-type chicken SERCA2a and SERCA2b, as well as their myc -tagged derivatives (Fig. 2A). mAb 9E10 recognized the human c-myc epitope that had been added to the carboxyl termini of SERCA2a and SERCA2b (Fig. 2B). Fig. 2B demonstrates that the correct microsomal preparations have expressed the myc -tagged ATPases and that wild-type SERCA2s do not react with mAb 9E10. The slight difference in molecular weights between SERCA2a-myc and SERCA2b-myc can be detected as a difference in electrophoretic mobility (Fig. 2B, lanes 2 and 4).
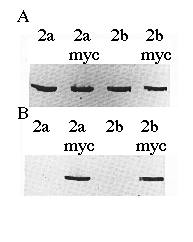
FIG. 2. Immunoblots of microsomes made from cells transfected with cDNA encoding SERCA2a, SERCA2a-myc, SERCA2b, or SERCA2b-myc as indicated. The proteins were detected with avian SERCA2-specific mAb 3H2 (panel A ) or human c-myc -specific mAb 9E10 (panel B). Equal amounts of protein (15 µg/lane) were loaded in each lane.
Epitope tags have been added to a variety of proteins in order to answer questions about topology and function. The human c-myc epitope has been used frequently for this purpose (Peculis and Gall, 1992; Ellison and Hochstrasser, 1991; reviewed in Kolodziej and Young (1991)). The caveat with epitope tagging of proteins is that the added epitope might disrupt native topology. If the topology were aberrant, then the misfolded protein would probably accumulate in the ER. For a protein which normally leaves the ER, subcellular localization of the tagged protein in the ER would reveal the misfolding. However, SERCA2 is an ER resident protein, so another assay was necessary to examine whether or not the added epitope disrupted the topology of SERCA2a-myc and SERCA2b-myc .
The best available assay to determine native conformation is a functional one. We measured the relative ability of the myc -tagged Ca2+-ATPases and wild-type Ca2+-ATPases to transport Ca2+. Microsomes were made from COS-1 cells after transfection with SERCA2a, SERCA2a-myc , SERCA2b, or SERCA2b-myc encoding cDNAs, and ATP-dependent uptake of 45Ca2+ was measured (see "Experimental Procedures"). The results (Fig. 3, A and B) show that Ca2+-ATPases with or without the c-myc epitope are equally competent to transport Ca2+. As a further test of native topology, thapsigargin, a potent inhibitor of both SERCA2a and SERCA2b (Campbell et al., 1991), was added to the reaction mixture. Both wild-type and myc -tagged Ca2+ATPases were inhibited by thapsigargin.
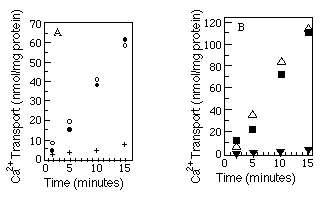
FIG. 3. Calcium uptake comparisons between tagged and
wild-type Ca2+-ATPases. Panel A compares wild-type SERCA2a
(![]() ) with SERCA2a-myc (O) and SERCA2a-myc preincubated
with thapsigargin (+). Panel B compares wild-type SERCA2b (
) with SERCA2a-myc (O) and SERCA2a-myc preincubated
with thapsigargin (+). Panel B compares wild-type SERCA2b (![]() )
with SERCA2b-myc (
)
with SERCA2b-myc (![]() ) and SERCA2b-myc preincubated
with thapsigargin (
) and SERCA2b-myc preincubated
with thapsigargin (![]() ). The myc -tagged data points
(
). The myc -tagged data points
(![]() and O) are the average of two independent experiments. Wild-type
and thapsigargin data points are each results of single experiments. 45Ca2+
uptake by microsomes made from mock transfected cells was always less than
10% of 45Ca2+ uptake by microsomes made from transfected
cells and has been subtracted from all the time points. The microsomes used
within a panel were prepared on the same day from cells transfected on the
same day.
and O) are the average of two independent experiments. Wild-type
and thapsigargin data points are each results of single experiments. 45Ca2+
uptake by microsomes made from mock transfected cells was always less than
10% of 45Ca2+ uptake by microsomes made from transfected
cells and has been subtracted from all the time points. The microsomes used
within a panel were prepared on the same day from cells transfected on the
same day.
Having shown SERCA2a-myc and SERCA2b-myc to be functionally unaltered, we addressed the question of topology using immunocytochemical techniques. The initial objective was to make the plasma membrane permeable to antibodies without perturbing the membrane of the ER. To achieve this, we utilized the bacterial cytolytic protein SLO. SLO is a 69-kDa protein which binds to cholesterol in a concentration-dependent manner. When mammalian cells were treated so that their cholesterol content fell by 50%, the cells became resistant to high concentrations of SLO (Duncan and Buckingham, 1980). Since the cholesterol concentration of the plasma membrane is 36 times higher than that of the ER (Keenan and Morré, 1970; Colbeau et al., 1971), it is not surprising that SLO was unable to permeabilize the ER sufficiently to allow antibodies access to the ER lumen (see below). To further ensure no internal membranes were permeabilized by SLO, a two-step procedure was employed (see "Experimental Procedures"). With high concentrations of SLO, lesions of about 13 nm can be produced in the plasma membrane (Buckingham and Duncan, 1983) through which antibodies can pass. Using this approach, the topology of SERCA2a-myc and SERCA2b-myc could be immunologically determined in whole cells with their ER membranes left intact.
Cells transfected with SERCA2a-myc or SERCA2b-myc encoding cDNAs were fixed, permeabilized with SLO or SLO plus saponin, which will permeabilize the ER as well as the plasma membrane. The cells were stained with mAb 9E10 (anti-myc ) or mAb 3H2 (anti-SERCA2) as outlined under "Experimental Procedures." Using SLO and mAb 9E10 without saponin, cells expressing SERCA2a-myc showed an intense staining around the nucleus (out of the focal plane) and a reticular/punctate staining pattern throughout the cell (Fig. 4a) that is characteristic of the ER (Louvard et al., 1982). However, there was no specific staining in similarly treated cells transfected with SERCA2b-myc cDNA (Fig. 4b). To verify that mAb 9E10 was able to label comparable levels of SERCA2b-myc and SERCA2a-myc , simultaneously transfected cells were permeabilized with SLO and saponin to reveal all c-myc epitopes (Fig. 4, c and d). There are about 105 cells on each coverslip, and our transfection efficiency is about 10%. In all cases where antibody labeling was positive, there were about 10,000 positively stained cells. However, on coverslips with SERCA2b-myc -transfected cells treated with SLO and mAb 9E10 without saponin (Fig. 4b), there were zero positively stained cells. This experiment has been repeated three times. Therefore the carboxyl terminus of SERCA2a-myc is cytosolic while that of SERCA2b-myc is lumenal.
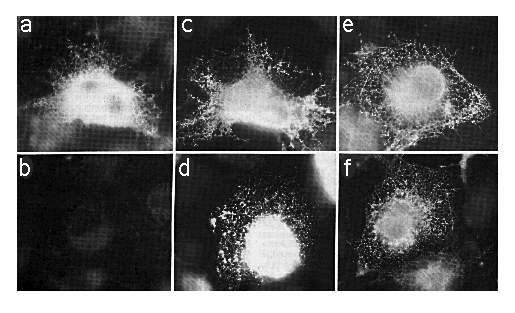
FIG. 4. Immunofluorescence photomicrographs of COS-1 cells transfected with cDNA encoding SERCA2a-myc (panels a, c, and e) or SERCA2b-myc (panels b, d, and f). Panels a and b were permeabilized with SLO alone and stained with mAb 9E10. Panels c and d were stained with mAb 9E10 after permeabilization with SLO and saponin. Panels e and f were permeabilized with SLO alone and stained with mAb 3H2. The exposure time for panel B was identical to that of panel A. All photomicrographs were taken with the x 63 objective lens and printed to the same magnification.
Additional controls were performed to confirm that the ER was not permeabilized by SLO. The c-myc epitope was placed on the lumenal domain of chicken Na/K-ATPase ß2-subunit.4 There was no intracellular mAb 9E10 staining in ß2-myc transfected cells permeabilized with SLO alone, but intense ER staining was present in transfected cells permeabilized with saponin (data not shown). This verified that mAb 9E10 has access to the ER lumen when saponin is used for permeabilization but not SLO. As another test, we employed a mAb to an epitope of rat dipeptidyl peptidase IV (Bartles et al., 1985) with a lumenal orientation. When COS-1 cells were transfected with dipeptidyl peptidase IV cDNA (Ogata et al., 1989) and immunofluorescently stained, there was no intracellular labeling in SLO-permeabilized cells but intense ER labeling of saponin-permeabilized cells was present.
We chose saponin to permeabilize the internal membranes because SLO and saponin (see Birk and Peri (1980)) are both cholesterol-specific. It is unlikely, therefore, that saponin would alter protein conformation and thus artifactually expose SERCA2a-myc or SERCA2b-myc epitopes. To confirm this, Ca2+dependent ATPase measurements (Lin and Morales, 1977) were conducted on rabbit light SR in the presence or absence of the same concentrations of saponin as used in Fig. 4, c and d. The ATPase activity of the Ca2+ATPase is retained in the presence of saponin. Similar results were obtained using C12E8 (data not shown; de Foresta et al., 1989). Therefore, it is very unlikely that the myc epitope on SERCA2b-myc was in the cytosol but inaccessible to mAb 9E10 (Fig. 4b) and became more accessible in the presence of saponin (Fig. 4d). Fig. 4, e and f, shows cells treated with SLO in the absence of any detergent and stained with mAb 3H2 to demonstrate cytosolic localization of the mAb 3H2 epitope, which is between amino acids 200 and 260 of SERCA2.
Why evolution has resulted in two similar forms of SERCA2 is not yet understood. The different topologies of SERCA2a and SERCA2b suggest a model to differentially regulate the two Ca2+-ATPases. Perhaps one of the many Ca2+ binding proteins of the ER acts as an accessory factor and interacts with the lumenal carboxyl terminus of SERCA2b. This would allow lumenal Ca2+ concentration-dependent regulation of SERCA2b. Furthermore, this mechanism would enable an individual cell, which expresses both SERCA2a and SERCA2b, to regulate the two Ca2+ATPases differentially. An example of such cells are cerebellar Purkinje cells which Plessers et al. (1991) have shown predominantly express SERCA2b but also express SERCA2a.
We have demonstrated that each form of SERCA2 has its carboxyl terminus in a different subcellular environment. Functional comparisons of SERCAs conducted in tissue-cultured COS cells (Campbell et al., 1991; Lytton et al., 1991) where the postulated regulatory factor(s) may not exist have revealed no differences among the various forms. It has been demonstrated that different tissues preferentially express either SERCA2a or SERCA2b (Lytton and MacLennan, 1988; Plessers et al., 1991; Campbell et al., 1991) so future studies might focus on tissues and cell types which may contain the hypothesized regulatory factors (e.g. brain). If regulatory factors were discovered, then we would finally begin to understand the selective pressures which resulted in distinct Ca2+ stores and Ca2+-ATPases to be highly conserved in birds and mammals. Nevertheless, we now have the first clue to functional and evolutionary differences of the nearly identical Ca2+-ATPases: SERCA2a and SERCA2b.
Acknowledgments - We thank Drs. Merianne Dieckmann (Stanford University) and Atsushi Miyajima (DNAX, Palo Alto) for kindly supplying COS-1 cells and the pcDL-SRa296 expression vector; Yutaka Sagara and Dr. Giuseppe Inesi for their help with the Ca2+-ATPase assays; Dr. Richard Pagano for guidance with the SLO procedure; Dr. Ann Hubbard for the dipeptidyl peptidase cDNA and antibody; Drs. Brenda Peculis and Joe Gall for the mAb 9E10 hybridoma; and Vic Lemas for the ß2-myc cDNA. We would also like to thank Delores Somerville for technical support.
* This work was supported by National Institutes of Health
Grants HL-02379 (to P. D. K.), GM-07231 (to A. M. C., predoctoral training),
HL-27867, and NS-23241 (to D. M. F.). The costs of publication of this article
were defrayed in part by the payment of page charges. This article must
therefore be hereby marked "advertisement" in accordance
with 18 U.S.C. Section 1734 solely to indicate this fact.
# Recipient of a clinician scientist career development award
from The Johns Hopkins University School of Medicine.
Footnotes
1 The nomenclature used here was adopted from Burk et al.
(1989).
2 The abbreviations used are: SLO streptolysin-O, COS-1, monkey
kidney tumor cell line transformed with SV40 large-T antigen; EGTA, [ethylenebis(oxyethylenenitrilo)]tetraacetic
acid; ER, endoplasmic reticulum- HEPES, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic
acid; mAb, monoclonal antibody; MOPS, 4-morpholinepropanesulfonic acid;
SR, sarcoplasmic reticulum.
3 PCR, polymerase chain reaction, is patented by Perkin-Elmer
Cetus.
4 V. Lemas, K Takeyasu, and D. Fambrough, manuscript in preparation.
REFERENCES
Bartles, J. R., Braiterman, L. T., and Hubbard, A. L. (1985) J. Biol.
Chem. 260, 12792-12802
Birk, Y., and Peri, I. (1980) in Toxic Constituents of Plant Foodstuffs
(Leiner, I. E., ed) 2nd Ed., pp. 161-182, Academic Press, Orlando, FL
Brandl, C. J., Green, N. M., Korczak, B., and MacLennan, D. H. (1986) Cell
44, 597-607
Buckingham, L., and Duncan, J. (1983) Biochim. Biophys. Acta 729,
115-122
Burk, S. E., Lytton, J., MacLennan, D. H., and Shull, G. E. (1989) J.
Biol. Chem. 264,18561-18568
Campbell, A. M., Kessler, P. D., Sagara, Y., Inesi, G., and Fambrough, D.
M. (1991) J. Biol. Chem. 266, 16050-16055
Clarke, D. M., Loo, T. W., and MacLennan, D. H. (1990) J. Biol.
Chem. 265, 17405-17408 \
Colbeau, A., Nachbaur, J., and Vignais, P. M. (1971) Biochim. Biophys.
Acta 249, 462-492
de Foresta, B., le Maire, M., Orlowski, S., Champeil, P., Lund, S., Moller,
J., Michelangeli, F., and Lee, A. G. (1989) Biochemistry 28,
2558-2567
Duncan, J., and Buckingham, L. (1980) Biochim. Biophys. Acta 603,
278-287
Eggermont, J. A., Wuytack, F., Verbist, J., and Casteels, R. (1990) Biochem.
J. 271, 649-653
Ellison, M. J., and Hochstrasser, M. (1991) J. Biol. Chem. 266,
21150-21157
Evan, G., Lewis, G., Ramsay, G., and Bishop, J. M. (1985) Mol. Cell.
Biol. 6, 3610-3616
Gravotta D., Adesnik, M., and Sabatini, D. D. (1990) J. Cell Biol. 111,
2893-2908
Gunteski-Hamblin, A.-M., Greeb, J., and Shull, G. E. (1988) J. Biol.
Chem. 263, 15032-15040
Kaprielian, Z., and Fambrough, D. M. (1987) Dev. Biol. 124,
490503
Karin, N. J., Kaprielian, Z., and Fambrough, D. M. (1989) Mol. Cell.
Biol. 9, 1978-1986
Keenan, T. W., and Morré, D. J. (1970) Biochemistry 9,
19-25
Kolodziej, P. A., and Young, R. A. (1991) Methods Enzymol. 194,
508-519
Kyte, J., and Doolittle, R. F. (1982) J. Mol. Biol. 157,105132
Laemmli, U. K. (1970) Nature 227, 680-685
Lin, T., and Morales, M. (1977) Anal. Biochem. 77, 10-17
Louvard, D., Reggio, H., and Warren, G. (1982) J. Cell Biol. 92,
92107
Lytton, J., and MacLennan, D. H. (1988) J. Biol. Chem. 263,
1502415031
Lytton, J., Westlin, M., and Hanley, M. R. (1991) J. Biol. Chem. 266,
17067-17071
MacLennan, D. H., Brandl, C. J., Korczak, B., and Green, N. M. (1985) Nature
316, 696-700
Matthews, I., Sharma, R. P., Lee, A. G., and East, J. M. (1990) J. Biol.
Chem. 265, 18737-18740
Ogata, S., Misumi, Y., and Ikahara, Y. (1989) J. Biol. Chem. 264,
3596-3601
Peculis, B. A., and Gall, J. G. (1992) J. Cell Biol. 116,
1-14
Plessers, L., Eggermont, J. A., Wuytack, F., and Casteels, R. (1991)
J. Neurosci. 11, 650-656
Takebe, Y., Seiki M., Fujisawa, J.-I., Hoy, P., Yokota, K., Arai, K.I.,
Yoshida, M., and Arai, N. (1988) Mol. Cell. Biol. 8, 466472
Wuytack, F., Eggermont, J. A., Raeymaekers, L., Plessers, L., and Casteels,
R. (1989) Biochem. J. 264, 765-769
![]()