Using Comparative Genomic Hybridization to Analyze Aneuploidy in Yeast
| We utilized Comparative Genomic Hybridization (CGH) in our attempts to characterize aneuploidy in Saccharomyces Cerevisiae. Two strains of yeast, α1 and α4, were grown for 2,000 and 5,000 generations under glucose limited conditions. (All evolved strains came from Dr. Clifford Zeyl of Wake Forest University). These evolved strains were compared with wild-type WTS288C. Below are scanned microarray slide images for all the crosses actually performed. In examining these images, keep in mind that WTS288C only received 70% of the recommended volume of Cy3 and Cy5 dyes, whereas the evolved strains received 100%. |
|---|
1. WTS288C v. WTS288C
|
2. WTS288C v. α12K
|
3. WTS288C v. α15K
|
4. WTS288C v. α42K
|
5. WTS288C v. α12K
|
6. WTS288C v. α15K
|
7. WTS288C v. α45K
|
8. WTS288C v. α45K |
9. WTS288C v. α42K |
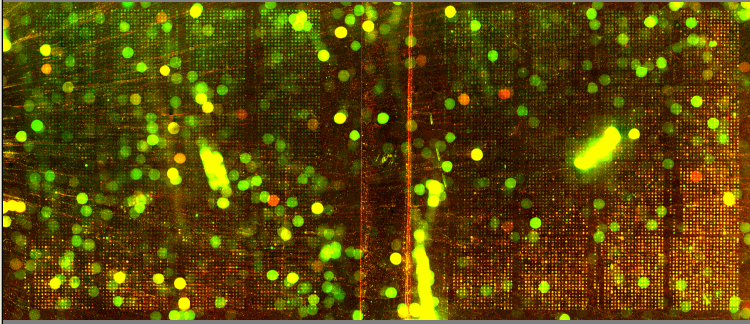
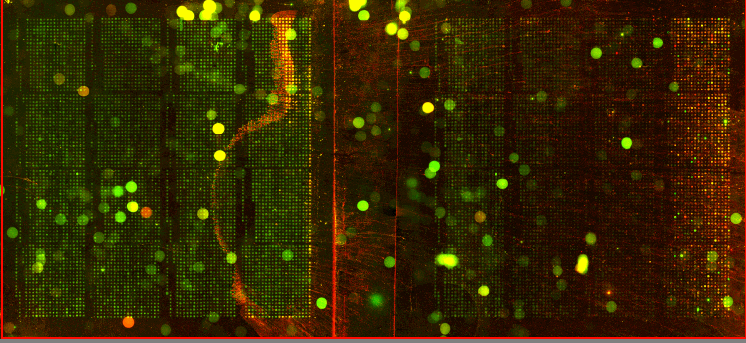

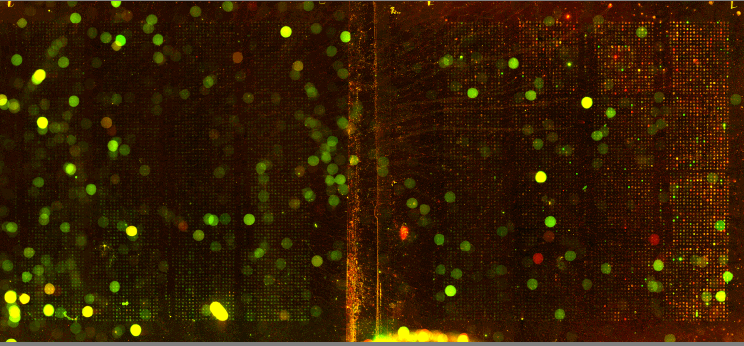

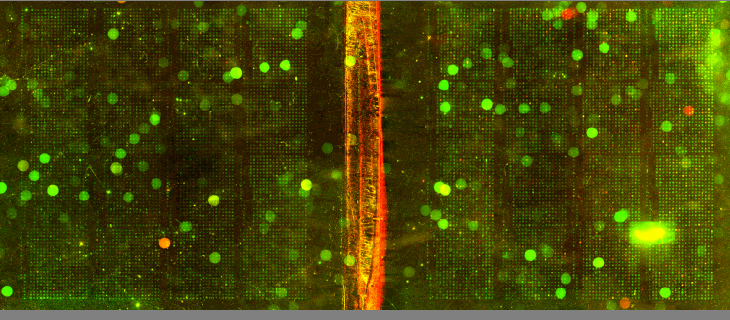
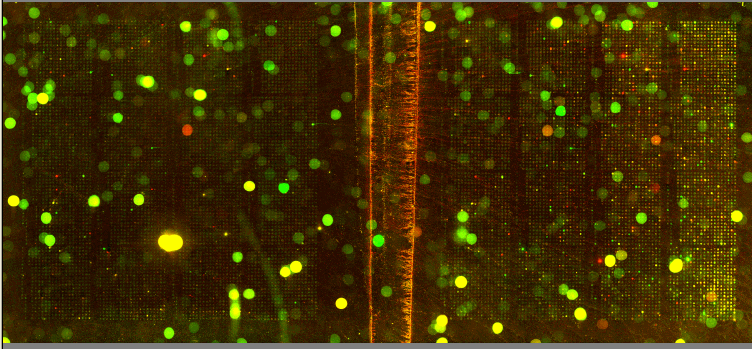

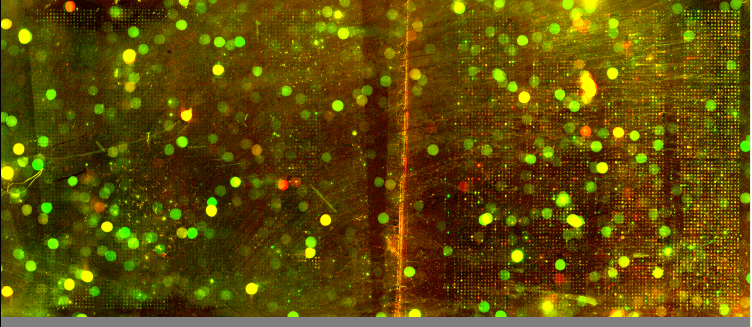
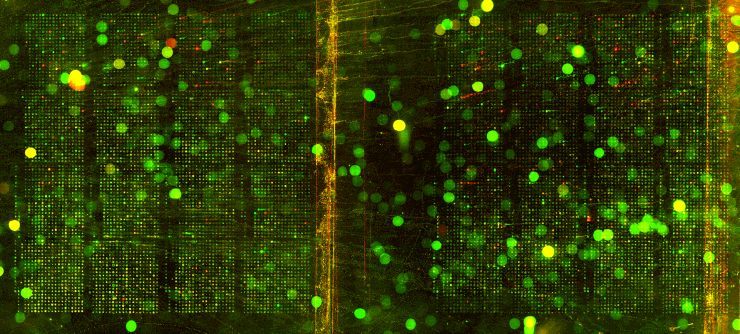

We based our CGH procedure off of A. Jacqueline Ryan's protocol and have the following suggestions for optimization. Due to limited amounts of dTTP and also Terminal
Deoxynucleotidyl Transferase, we were unable to tag α45K and α42K,
eliminating crosses 8 and 9.
Low Signal The intensity of dendrimer signal on the microarray slides was lower than expected for the initial slides hybridized. Low concentration of DNA was hypothesized to be an explanation of the low intensity. We nanodropped DNA about to be used for hybridization that had already been tagged and purified (DNA had gone through step 48 in Ryan's protocol). Results showed we had less DNA to hybridize than anticipated (Table 1). Table 1: Spectrophotometry data from two crosses.
These data reveal we had around 5 of the 10
/μg of DNA expected.
Concentrations listed are the result of a 10-fold dilution. We had
a total volume of 10 μL per cross.
We have a lot of background noise and dust particles on the slide images. Our adjustments to the contrast setting emphasized background signal more noticeably than had the signals been more intense. We planned to use MAGIC Tool to produce Cy3/Cy5 ratios for the spots, but did not for several reasons. It was difficult to see the DNA spots well enough to put a grid around them in the earlier versions of MAGIC Tool (the latest version probably would have worked). The color and intensity of spots varied qualitatively between the two identical halves on the same slide (slides 1-7). Additionally, our control slide WTS88C v. WTS288C did not produce the predicted yellow spots, but rather varied in color between red, yellow, and green depending on the general region of the slide (slide 1). Brief Discussion and Recommendations Somewhere between step 18 and step 48 of Ryan's protocol, we lost half of our DNA. All of the strains were nanodropped after step 18 and although only two of the crosses were nanodropped after step 48, these probably were representative since all the DNA was processed together. Retrospectively, we should have fragmented, tagged, and purified DNA (steps 18-48) for a smaller quantity of slides. Dr. Campbell made this suggestion afterwards upon thinking of ways we may have conducted this experiment differently. We attempted to process DNA for 14 slides at one time. The purification steps were cumbersome. We were going through the protocol for the first time with a large quantity of DNA. Our procedure differed from the written protocol in many seemingly minor ways which may have affected our results. We should have amended the digestion times in the tagging step to account for the 10-fold increase in volume. More time may have enabled the enzymes to be more effective. Incubation times, however, could not explain why the DNA concentration had halved, and we have yet to find a satisfying explanation. We highly recommend doing steps 1-18 in batch style to save time. Our results after step 10 had especially high quality (260/280 of ~1.9) and decent concentration. After step 18 our results had less quality (260/280 of ~1.66), but still adequate concentration. Because of the quantified success in the initial 18 steps, it seems logical to continue to perform these steps in batch style. Best wishes for successful CGH! Acknowledgements: We would like to thank Dr. Campbell for his guidance, A. Jacqueline Ryan for her protocol and recommendations, and last year's Genomics class for their papers guiding us in the RTPCR process. |
|---|