
This web page was produced as an assignment for an undergraduate course at Davidson College.
Cancer Genomics
3D structure of HER2 Kinase Domain. Image Courtesy of PBD (Permission Pending).
Cancer Genomics and Classification
Cancer is responsible for one in eight deaths across the world (Stratton, 2009). Its high prevalence coupled with high diversity makes it a pervasive, complex issue. Cancer genomics seeks to identify and understand the genomic changes that trigger the switch from normal cell to cancerous cell.
Cancer can be divided into three types.
1. Familial cancer accounts for 10-15% of all cancer cases and is characterized by the same type of cancer appearing in two or more relatives from the same family line.
2. Hereditary cancer accounts for 5-10% of cancers and stems from a single gene mutation in a parental germline.
3. Sporadic cancer is the most common, accounting for 75% of all cases. This type involves mutations due to nonhereditary factors (Santos, 2013).
Types of Tumor Mutations
Not all cancers contain the same mutations. Not even all cancers of the same type contain the same mutations. This makes it especially difficult to treat because the mutations present may affect how a tumor responds to treatment (Santos, 2013).
Cancer is divided into four levels of heterogeneity.
1. Intratumoral refers to the heterogeneity within one tumor.
2. Intermetastatic refers to heterogeneity across multiple metastases in one patient. Each mestastasis forms from a primary tumor cell that multiplies in a suitable environment elsewhere in the body.
3. Intrametastatic refers to heterogeneity in a single metastasis. This type is much like intratumoral but refers to a cluster of cells that grow independent of the primary tumor. We can say that metastasis heterogeneity is two-fold. The founding metastatic cell has a subset of mutations that accumulated during the formation of the primary tumor. Subsequent metastatic cells accumulate more mutations as division occurs.
4. Finally interpatient refers to heterogeneity among different patients' tumors.
Two types of mutations occur within tumors. Driver gene mutations occur in a subset of "cancer genes" (Stratton, 2009). They are selected for during development, because they provide a growth advantage to the cell. The advantage is normally on the order of a 0.4% increase. Passenger gene mutations occur randomly throughout all genes and do not confer a selective growth advantage.
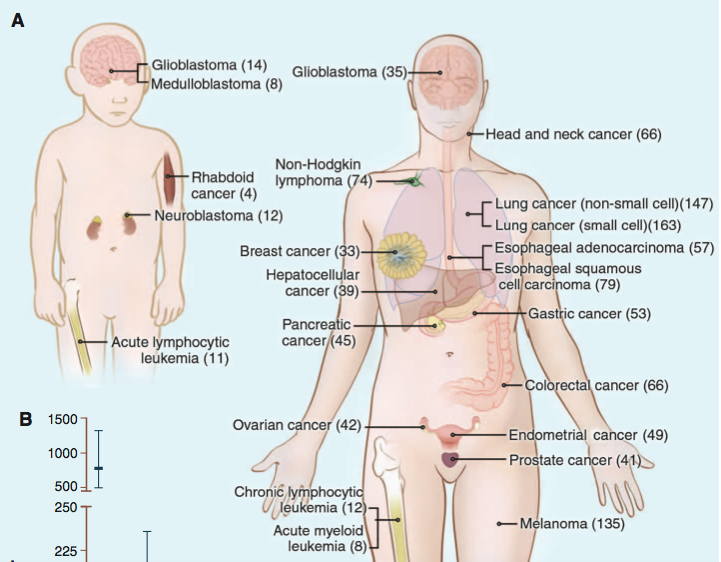
The average tumor contains 33-60 somatically mutated genes that may alter protein function (Fig. 1). About 95% of somatic mutations are single-base substitutions. Broken down further, 90.7% of these are missense, 7.6% are nonsense, and 1.7% are splice or UTR mutations. The remaining 5% of somatic mutations are insertions or deletions (Vogelstein, 2013). About 90% of somatic mutations are dominant (Stratton, 2009).
Fig. 1 Number of somatic mutations in representative human cancers, detected by genome-wide sequencing studies. (A) The genomes of a diverse group of adult (right) and pediatric (left) cancers have been analyzed. Numbers in parentheses indicate the median number of nonsynonymous mutations per tumor.
(B) The median number of nonsynonymous mutations per tumor in a variety of tumor types. Horizontal bars indicate the 25 and 75% quartiles. MSI, microsatellite instability; SCLC, small cell lung cancers; NSCLC, non-small cell lung cancers; ESCC, esophageal squamous cell carcinomas; MSS microsatellite stable; EAC, esophageal adenocarcinomas. The published data on which this figure is based are provided in table S1C.
Image Courtesy of Vogelstein B et al. 2013. Cancer Genome Landscapes. Science 339: 1546-58. (Permission Pending)
Keep in mind that tumor point mutation rates are similar to rates found in normal cells. The main difference lies in the cell's ability to detect and correct mutations as well as in the rate of chromosomal changes. Tumor cells have a higher rate of chromosomal insertions, deletions, and translocations than that of normal cells. Translocation breakpoints often occur in so-called "gene deserts" which are regions without any known genes (Vogelstein, 2013). Sometimes translocations create fusion genes or move genes closer to regulatory elements, both of which can lead to abnormal gene expression (Stratton, 2009).
Cancer also contains epigenetic and transcriptional mutations, but these types are not as easily studied and thus have not been the focus of most cancer genomics research endeavors (Chin, 2011).
Databases and Their Application
The Cancer Genome Atlas (TCGA) began in 2006 as a coordinated pilot program by the National Cancer Institute and National Human Genome Research Institute to classify the genomic changes in over 20 types of cancer. The program proved a useful and freely available tool for researchers and has since been extended to classify an additional 20 tumor types. The cancerous tissues being studied include breast, CNS, endocrine, gastrointestinal, gynecologic, head and neck, hematologic, skin, soft tissue, thoracic, and urologic. Their sequence, alignment, and mutation data is stored as the Cancer Genomics Hub (CGHub), which is operated by the University of Santa Cruz.
The International Cancer Genome Consortium (ICGC) seeks to classify genomic changes in 50 types and subtypes of cancer. It aims to coordinate cancer genomics research efforts and make the data readily accessible to researchers. Some of the cancers being studied include biliary tract, breast, brain, bone, cervical, esophageal, eye, liver, lung, pancreatic, prostate, renal, skin, and thyroid. The ICGC data portal releases data from each of its research projects on a quarterly basis.
At the University of Michigan, the Michigan Center for Translational Pathology demonstrated how genomic data in databases like TCGA and ICGC can be used to treat cancer patients. The Michigan Oncology Sequencing Project (MI-ONCOSEQ) aimed to use whole-genome, whole-exome, and transcriptome sequencing to place patients into clinical trials that were most relevant to their tumor's particular mutations. They first tested their technique on xenografts from prostate cancer patients and then tested it on two patients, one with metastatic colorectal cancer and the other with malignant melanoma.
Application to Personalized Medicine
The long-term goal of data collection is to use genomic information about a patient's tumor to develop personalized treatment options. Physicians can better select drugs to target tumor cells if they know which mutations are present in a patient's tumor. For example, microarray analysis and immunohistochemistry can distinguish between different types of breast cancer. HER2-positive tumors are treated with trastuzumab while HER2-negative tumors are treated with adjuvant chemotherapy (Santos, 2013). Personalized medicine based in genomics may also protect patients from undergoing treatments that will not effectively attack the tumor. Only lung cancer patients with an EFGR mutation or ALK translocation respond to the drugs that are currently available on the market.
Many cancer drugs are kinase inhibitors that impede enzyme function. Researchers need to develop drugs that target non-enzymatic functions and drugs that target restoring defective gene function. This is of course easier said than done (Vogelstein, 2013). As it stands now, cancer genomics is trending towards treating tumors based on biological behavior rather than anatomical location (Yousef, 2012). Most agree this trend is much needed.
References
Stratton MR, Campbell PJ, Futreal PA. 2009. The cancer genome. Nature 458: 719-24.
Vogelstein B et al. 2013. Cancer Genome Landscapes. Science 339: 1546-58.
Genomics Page
Biology Home Page
© Copyright 2014 Department of Biology, Davidson College, Davidson, NC 28035

{kind=link}