Gjoneska et al.
aimed to characterize the epigenetic changes which underlie
alterations in gene regulation resulting in neurodegeneration
associated with Alzheimer’s disease (AD). The researchers discuss
that much of this work has not been performed due to the
inaccessibility of human brain samples but solve this problem with
their model organism: CK-p25 mice, for which accumulation of the
p25 Cdk5 activator protein is inducible and which demonstrate an
AD-like phenotype. The researchers collected both transcriptomic
and epigenetic data to compare gene expression and chromatin
modification in CK-p25 mice and CK littermate controls at both
early and late stages of neurodegeneration (2 weeks and 6 weeks
following induction of p25 accumulation in CK-p25 mice). The
researchers sought to investigate possible mutations leading to
epigenomic modifications which may contribute to gene-regulatory
changes previously identified as associated with AD.
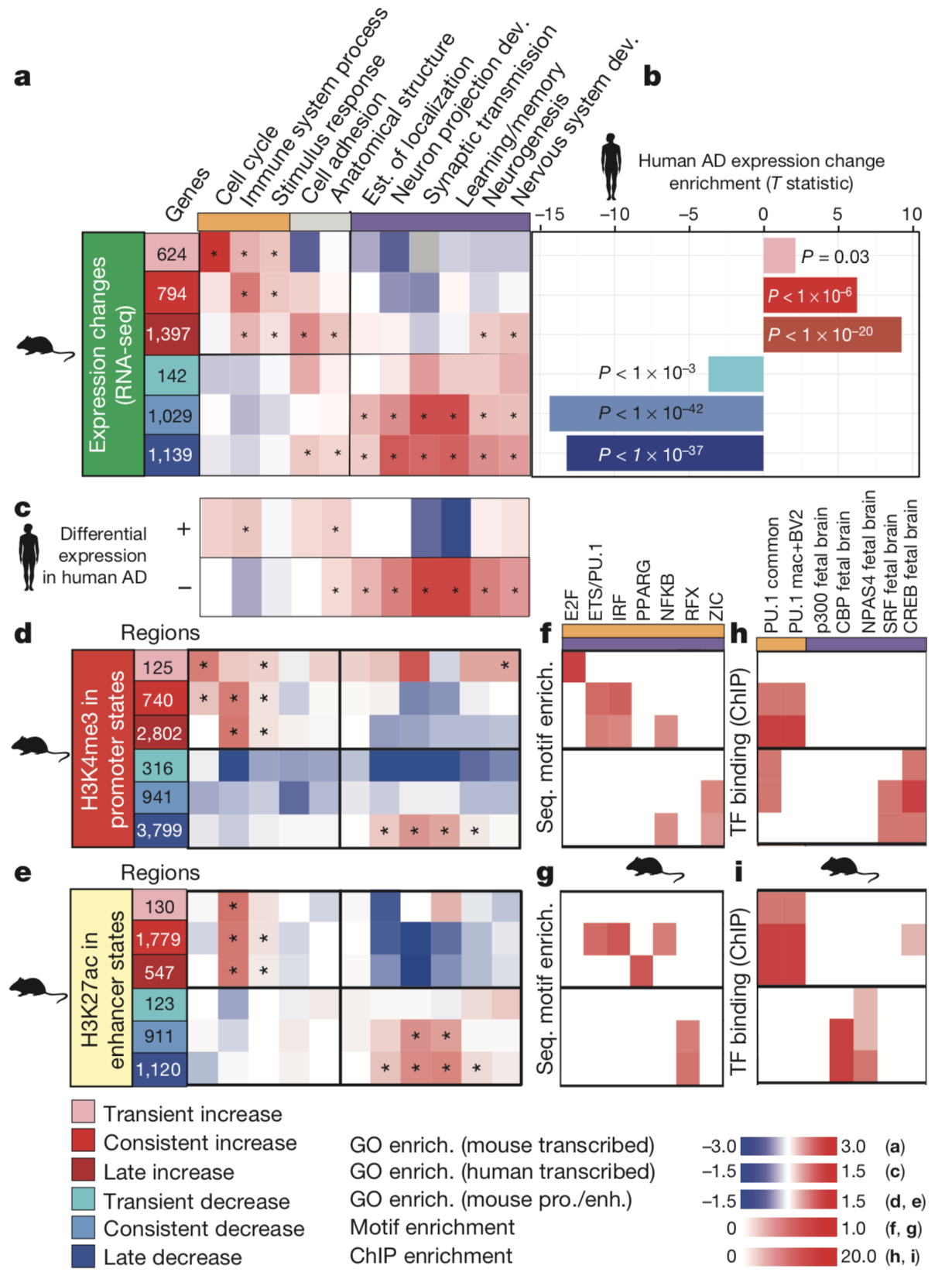
To analyze the transcriptome, the researchers performed
RNA sequencing at both the 2-week and 6-week time points and
characterized the upregulated and down regulated genes into three
categories: transient (differentially expressed only at the 2-week
time point), late-onset (only at the 6-week time point) or
consistent (differentially expressed at both time points). The
researchers analyzed common functions and gene ontology (GO) terms
associated with the genes in each category to possibly uncover
coordinated transcriptomic regulation of processes which may
contribute to the pathophysiology of AD.
To analyze the epigenome, the researchers employed
chromatin immunoprecipitation sequencing (ChIP-seq) to detect
seven chromatin marks throughout the genome, before focusing their
analysis upon H3K4me3 marks to identify active promoters within
promoter chromatin regions and H3K27ac marks to identify active
enhancer regions within enhancer chromatin states. The researchers
identified regions with increased or decreased levels of either
H3K4 trimetlylation (H3K4me3; promoter regions) or H3K27
acetylation (H3K27ac; enhancer regions). In addition, in a similar
fashion to their characterization of transcriptomic changes, the
researchers also profiled these altered-level epigenomic marks as
transient, late-onset, or consistent. Combining their temporal
observations with thorough, genome-wide epigenomic and
transcriptional profiling the researchers established sufficient
data from which they could draw conclusions about coordinated
changes in gene regulation to investigate the mechanisms of AD.
The researchers found consistent trends in gene ontology
analyses between their transcriptomic and epigenomic data. These
results included concordance of transcriptomic upregulation and
adjacent increased-level promoter or enhancer regions with genes
involved in immune and stimulus response functions among CK-p25
mice relative to CK littermate controls. Additionally, the results
indicated concordance of transcriptomic downregulation and
adjacent decreased-level promoter or enhancer regions with genes
involved in synapse and learning-associated functions.
Having observed transcriptomic and epigenetic changes
broadly, the researchers shifted their focus to the transcription
factors which bind the epigenetic regions they identified in order
to regulate transcription. The researchers found distinct and
consistent transcription factor motifs and binding patters for
both increased- and decreased-level regulatory regions.
Increased-level promoters and enhancers demonstrated binding of
transcription factors known to regulate genes involved in immune
functions. Conversely, decreased-level promoters and enhancers
demonstrated binding of transcription factors known to regulate
neuronal activity.
Next, the researchers assessed the reproducibility of
their observations in humans. In one experiment, the researchers
assayed the ability of human transcription factors orthologous to
those which bound to increased-level enhancer regions to drive in
vitro gene expression in mouse cell line models of
brain-specific immune cells and neuroblastoma cells. The data from
this experiment indicated the majority of human transcription
factors tested could drive in vitro gene expression. These
results demonstrate functional conservation of the increased-level
enhancer regions observed in AD mice.
The researchers next
examined a possible causal relationship between the changes in
regulatory regions they observed in CK-p25 mice and AD in humans.
The researchers first assessed the enrichment for human
AD-associated single nucleotide polymorphisms (SNPs) found in
genome-wide association studies (GWAS) in increasing-level and
decreasing-level regulatory regions and found significant
enrichment for AD-associated SNPs in both consistent- and
late-increasing enhancers, while promoters demonstrated only weak
enrichment at any time scale. These results suggest variants in
distal enhancer sequences, rather than in adjacent promoters, are
associated with AD predisposition.
The researchers then
narrowed the cell types in which these effects mediate AD
phenotypes by investigating concordance between enrichment for
human AD-associated SNPs and enrichment for consistently
increasing-level and consistently decreasing-level enhancer
orthologues in diverse human cell types and tissues. Consistently
increasing-level enhancers demonstrated a positive correlation
with human AD-associated SNPs (R2 = 0.49), but
consistently decreasing-level enhancers only showed a very weak
correlation (R2 = 0.05). It was apparent in
these data that human immune cell types demonstrated higher
enrichment for both human AD-associated SNPs and consistently
increasing-level enhancers than any other type of cells, including
neuronal cells. These results implicate that genetic
predisposition to AD is primarily attributable to immune
functions, rather than neuronal functions, which may be impacted
by other factors such as aging and environmental influences.
Finally, the
researchers turned their focus to GWAS-determined AD-associated
human genetic loci which lie in increased-level enhancer mouse
orthologs as potential candidates for further experiments. In one
such an experiment, the researchers demonstrate the ability for a
known, AD-associated SNP located in an enhancer region upstream of
the immune-function regulating transcription factor gene, SPI1,
to significantly amplify enhancer activity as compared to wild
type in an in vitro experiment employing mouse model
brain-specific immune cells. This result provides a direct
connection between AD-associated SNPs in enhancer regions and
genes regulating immune function. The study concludes by laying
out a model of AD in which genetically-driven immune
dysregulation–as this investigation suggests is mediated by
mutations in increased-level enhancer regions–combines with
environmentally-attributable epigenomic changes in neuronal cells
to enhance immune susceptibility to AD-associated environmental
factors during aging and cognitive decline.
This paper was a
well-written presentation of challenging concepts with which many
outside the field may not be familiar. Investigating epigenomic
mechanisms provides a challenge to biologists unlike
transcriptomic or genetic mechanisms because much less is known
about epigenomics than other, more well-studied fields. One way in
which the authors make epigenomics approachable is by collecting
their epigenomic data and presenting these data in parallel to
transcriptomic data. High throughput transcriptomic data, with
which genomicists are very familiar, are often presented as
heatmaps quantifying deviation of expression from that in a
control, just as they are in this paper. The researchers in this
study collected epigenomic data in a similar manner to that in
which transcriptomic data is collected, quantifying levels of
epigenomic marks relative to levels of these marks in controls.
The authors present the epigenomic data they collected in a
similar manner to their transcriptomic data, in heat maps which
conserve the color scale used when presenting their transcriptomic
data.
I particularly
appreciated the conservation of color legends the authors
preserved in all of the figures of the paper. Colors established
to determine upregulation, downregulation, timing, and cell types
in Figure 1 were preserved in Figures 2 and 3 and made drawing
meaningful conclusions from these figures easier. The figures were
presented in an intuitive manner and were well marked with symbols
denoting the source of data, either from human studies or from
mice.
Finally, the quality of science in this paper was elegant. The researchers are very thorough in their approach and take care to demonstrate the quality of their model organism, the Ck-p25 mouse, for studies investigating AD. Additionally, I appreciated the honesty of the researchers in telling the story of both the promoter regions and the decreased-level enhancers even though these stories do not feature prominently in their overall conclusion. Telling these stories allowed me to see the process by which they narrowed down the data to eventually come to determine the important role increased-level enhancers seem to play in AD.


- Gjoneska et al. (2015) Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer's disease Nature 518:365-369.
Email Questions or Comments: owkoucky@davidson.edu
© Copyright 2018 Department of Biology, Davidson College, Davidson, NC 28035