- This web page was produced
as an assignment for an undergraduate course at Davidson College -
- by Rebecca Jameson -
Rabies
Introduction
Pathogenesis
Epidemiology
Diagnosis/Treatment
References
Introduction
Rabies is a fatal central nervous system (CNS) disease responsible for approximately
60,000 annual deaths worldwide, making it the tenth most common lethal infectious
disease (Dietzschold et al. 2005). The causative agent is a neurotropic virus
consisting of nonsegmented, negative-stranded RNA contained within a bullet-shaped
envelope. Rabies virus (RV) is 1 of 7 serotypes belonging to the genus Lyssavirus
and the family Rhabdoviridae (Plotkin 2000). The most common site of RV entry
in humans is the skin or mucous membrane, where the virus is delivered into the
muscle and subcutaneous tissue through biting, licking or scratching by an RV-infected
animal (Warrell et al. 2004). Disease can present with one of two clinical forms.
In the majority of rabies cases, the pathologic manifestation in the CNS is acute
encephalomyelitis. This form is known as classic or encephalitic (furious) rabies
and comprises 80-85% of rabies cases. It is distinguished by neurotropism, neuroinvasiveness
and impaired neuronal functions (Jackson 2002). The ssymptoms of classic rabies
include hydrophobia, pharyngeal spasms, and hyperactivity leading to paralysis,
coma and death (Hankins et al. 2004). Paralytic rabies is a less common clinical
form characterized by the development of prominent and flaccid muscle weakness
(Jackson 2002). Death in both clinical and paralytic rabies ultimately results
from neuronal dysfunction due to the dramatically inhibited synthesis of proteins
required for maintaining neuronal functions (Dietzschold et al. 2005).
Rabies progresses through 5 clinical stages that can vary
depending on the extent of the bites, the amount of secretion, and the proximity
to the CNS, with disease transmitted through bites close to the brain progressing
more rapidly than disease transmitted through bites on the lower extremities
(Hankins et al. 2004). The incubation stage ranges from 10 days to 1 year, with
the average lying somewhere between 20-60 days. The prodrome stage occurs 2
to 10 days after exposure and can last from 1 day to 2 weeks. This stage is
characterized by nonspecific flu-like symptoms such as fever, malaise, headache
and nausea (Jackson 2002). Acute neurologic syndrome occurs 7-10 days after
the onset of prodrome and includes dysarthia, dysphagia, excessive salivation,
vertigo, agitation, visual and auditory hallucinations, hydrophobia secondary
to painful contractions of pharyngeal muscles, and polyneuritis. Coma occurs
7-10 days after the onset of acute neurologic syndrome. This stage includes
hydrophobia, prolonged apnea, generalized flaccid paralysis, seizures and coma
with acute respiratory collapse. Death may follow 2-3 days after the onset of
paralysis (Hankins et al. 2004).
Back to Top
Pathogenesis
The
viral particle is comprised of a membrane made of host lipids and two proteins,
G and M, surrounding a helical nucleocapsid (Thoulouse et al. 1998). As shown
in figure 1, the helical coil of the ribonucleoprotein(RNP) complex core is
formed by the rabies genome, a single non-segmented strand of negative-sense
RNA, a nucleoprotein, a phosphoprotein, and an RNA-dependent RNA polymerase.
The RNP core is covered by a layer of matrix protein, which is in turn covered
by a host-derived lipoprotein envelope studded with rabies glycoprotein (Warrell
et al. 2004). The trimeric G protein exposed on the surface of the virion is
responsible for the attachment to the target cell by interaction with several
cell membrane components. In particular, the nicotinic acetylcholine receptor
(nAChR) acts as a receptor for RV. Binding of RV to nAChRs localizes and concentrates
the virus on postsynaptic cells, which in turn facilitates subsequent uptake
and transfer of the virus to peripheral motor nerves (Jackson 2002). In neurons
that do not express nAChRs, it has been shown that other molecules such as phospholipids,
gangliosides, neuronal cell adhesion molecule and the nerve growth factor receptor
can serve as an RV receptors (Dietzschold et al. 2005). Following attachment
to the G protein to the cell membrane, RV enters the cell by endocytosis and
then resides in an early endosomal compartment. The acidic environment of the
endosome causes fusion of the viral membrane with the endosomal membrane (Jackson
2002). This allows for the nucleocapsid to escape into the cytosol, where transcription
and replication occur (Thoulouse et al. 1998).
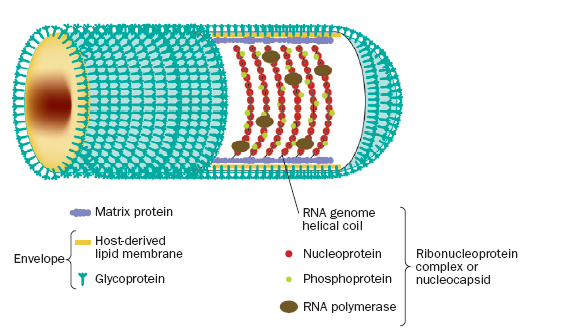
Figure 1.
The internal ribonucleoprotein (RNP) core of the rabies virion consists of a
negative-sense genome RNA encapsidated by nucleoprotein, polymerase cofactor
phosphoprotein, and the virion-associated RNA polymerase. The RNP core is covered
in matrix protein and surrounded by a lipid-bilayer envelope (Warrell et al.
2004). Permission to use figure pending.
Although RV can infect a variety of cell types, it
primarily targets neurons. The cycle of viral infection is depicted in figure 2. The virus spreads by retrograde axonal transport
from the peripheral nerves to the neuronal cell body, possibly by cytoplasmic
dynein (Wang et al. 2005). After replication in the cell body of the primary
neuron, infection proceeds via retrograde axonal transport and transsynaptic
spread through several neurons, as illustrated by figure 3. Transsynaptic spread
is the ability of the virus to use synaptic junctions to propagate within the
CNS (Dietzschold et al. 2005). Neuronal infection by RV causes abnormalities
in the function of neurotransmitters affecting serotonin, GABA and muscarinic
acetylcholine transmission (Warrell et al. 2004). Acinar cells are infected
next, which in turn shed the virus into the oral cavity. This accounts for the
presence of the virus in saliva (Dietzschold et al. 2005).

Figure 2. The cycle of rabies infection begins with viral entry at a peripheral site and proceeds through retrograde axonal transport. Viral replication occurs in the cell body of the primary neuron. Infection proceeds by transsynaptic spread through several neurons before spreading to the acinar cells, which then shed the virus into the saliva (Dietzschold et al. 2005). Permission to use figure pending.
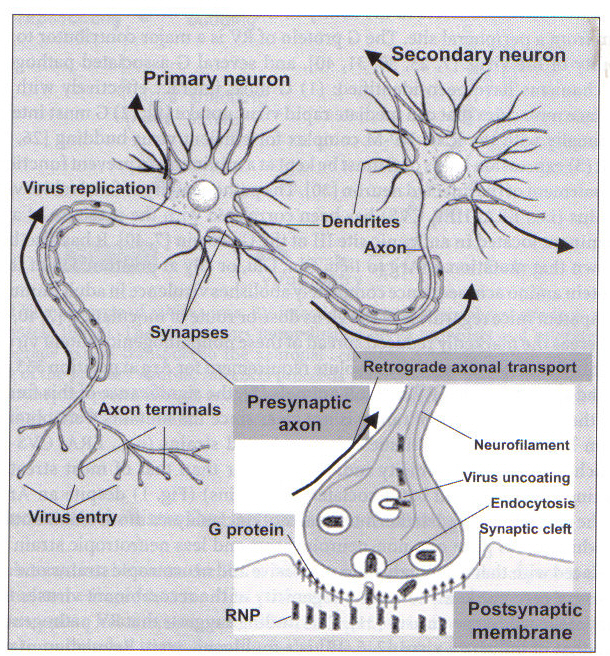
Figure 3. The neuroinvasiveness of the virus
results from its ability to migrate to the central nervous system (CNS) through
retrograde axonal transport and transynaptic spread. Rabies virus spreads from
the postsynaptic site to the presynaptic site via receptor-mediated endocytosis.
In retrograde axonal transport, the ribonucleoprotein complexes of the virus
are carried by direct attachment to a dynein motor or by encapsulation in vessicles
attached to a dynein motor (Dietzschold et al. 2005). Permission to use figure
pending.
Back to Top
Epidemiology
Because animal bite is the primary route of RV transmission
to humans, the epidemiology of human rabies is a direct reflection of the regional
animal reservoir for rabies virus and the opportunity for human-animal interaction
(Childs 2002). Worldwide, canid species are the main vector in the transmission
of rabies to humans, particularly in developing countries where canine rabies
is endemic (Krebs 1995). In countries where canine rabies persists, the age
and sex distribution of human rabies deaths generally mirrors the distribution
of dog bit victims. The Indian subcontinent, Southeast Asia and most of Africa
are the major foci of rabies today, with more than 30,000 cases each year in
India alone (Plotkin 2000). Crowded urban centers with inadequate public health
infrastructures are prone to transmission of rabies virus. In developed countries
where canine rabies has retreated, the transmission of rabies by wild mammals
accounts for 90% of human exposures (Hankins et al. 2004). Rabid bats, especially
silver-haired bats, are the most prevalent source of human rabies in the United
States. Between 25,000 and 40,000 people in the US are treated annually for
exposure to rabid or potentially rabid animals (Hankins et al. 2004).
Other rare routes of rabies transmission include handling of infected carcasses, consumption of raw, infected meat and inhalation of aerosolized rabies in caves inhabited by millions of bats. At least eight cases of human-to-human transmission of rabies have resulted from the transplantation of infected corneas (Jackson 2002).
Back to Top
Diagnosis/Treatment
Diagnosing rabies can be difficult if no history of
exposure reported, and it is often misdiagnosed as Guillain-Barre syndrome,
poliomyelitis and other viral encephalitides, or laryngeal disorder (Plotkin
2000). A conclusive rabies diagnosis can be made in humans before death by observing
virus-specific fluorescent material in skin biopsy specimens, isolating the
virus from patient saliva, or detecting the presence of antirabies antibodies
in the serum or cerebrospinal fluid of patients who have not been immunized
(Hankins et al. 2004). The basic principles behind rabies prophylaxis are the
removal of free virus from the body by both washing and neutralization, followed
by the induction of a rabies virus-specific immune response in the exposed individual
before rabies virus can replicate in the CNS. This requires the administration
of both passive and active vaccination. In passive vaccination, rabies immune
globulin (RIG) from adults who have been immunized with rabies vaccine is administered
to previously unimmunized people so as to passively impart antibodies. There
are three primary types of active rabies vaccinations currently administered
to humans throughout the world: nerve tissue-derived vaccines (NTVs), high-quality
cell culture vaccines produced under stringent quality control and lower-quality
cell culture vaccines that do not adhere to FDA regulations of national pharmacopeia
standards in industrialized countries (Briggs et al. 2002). NTVs are produced
from brain tissue of animals infected with a fixed strain of rabies virus. After
the brain tissue is harvested, the virus is inactivated and then diluted to
a concentration of 2-5% of brain tissue. NTVs are extremely painful, however,
and can cause severe neurologic adverse reactions due to the presence of myelinated
tissue in the vaccine (Briggs et al. 2002). Unfortunately, nerve tissue vaccines
are still the most widely used prophylaxis for rabies. The optimal rabies
vaccine today is human diploid cell vaccine (HDCV), which is a type of cell-culture
vaccine produced in human fibroblasts. Unfortunately, treatment is generally
unsuccessful when administered after the patient becomes symptomatic. Therefore,
efforts must be focused on disease-preventing measures (Vanniasinkam et al.
2004).
Back to Top
References
Childs, JE. (2002). Epidemiology. In: Rabies (eds. AC Jackson and WH Wunner).
Academic Press, 493.
Dietzschold, B, M Schnell, H Koprowski. (2005). Pathogenesis of Rabies. Current
Topics in Microbiology and Immunology. 292: 45-56.
Hankins, DG. (2004). Overview, prevention, and treatment of rabies. Mayo Clinic
Proceedings. 79(5): 671-6.
Jackson, AC. (2002). Human disease. In: Rabies (eds. AC Jackson and WH Wunner).
Academic Press, 493.
Janeway, A.C, P. Travers, M. Walport., J.M. Shlomchik.2005. Immunobiology:
The Immune System in Health and Disease. 6th ed. New York, NY: Garland
Publishing. pp 196, 416-417.
Krebs, JW, ML Wilson, JE Childs. (1995). Rabies: epidemiology, prevention and
future research. Journal of Mammalogy. 76(3): 681-94.
Plotkin, SA. (2000). Rabies. Clinical Infectious Diseases. 30(1): 4-11.
Thoulouse, MA, M Lafage, M Schachner, U Hartmann, H Cremer, M Lafon. (1998).
The Neural Cell Adhesion Molecule Is a Receptor for Rabies Virus. Journal of
Virology. 72(9): 7181-7190.
Vanniasinkam, T and HCJ Ertl. (2004). Rabies vaccines: the third generation.
Letters in Drug Design and Discovery. 1: 289-292.
Wang, ZW, L Sarmento, Y Wang, X Li, V Dhingra, T Tseggai, B Jiang, ZF Fu. (2005).
Attenuated rabies virus activates, while pathogenic rabies virus evades, the
host innate immune responses in the crentral nervous system. Journal of Virology.
79(19): 12554-12565.
Warrell, MJ and DA Warrell. (2004). Rabies and other lyssavirus diseases. The
Lancet. 363(9413): 959-69.
Back to Top
Rebecca's Homepage
Immunology Homepage
Davidson
College Homepage
Send comments, questions, and suggestions to: Rebecca Jameson
© Copyright 2006 Department of Biology, Davidson College, Davidson,
NC 28036