Figure 1. Morphea Note the discolored plaques (courtesy of dermnet.com Scleroderma).
*This web page was produced as an assignment for an undergraduate course at Davidson College *
Basic Information
Scleroderma is an autoimmune disease, most often recognized by the presence of hard, rough skin. The actual term “scleroderma” comes from two Greek words, “skleros” and “derma,” meaning “hard skin” (Jimenez et al., 2006).
There are two forms of scleroderma, localized and systemic. Localized scleroderma has three subtypes: morphea, generalized morphea, and linear scleroderma. Morphea typically involves plaques on the skin with ivory coloration, usually in just one area of the body. Generalized morphea is more severe, involving more areas of the body, higher instances of plaques and lesions, and often the patient also shows hyperpigmentation. Linear scleroderma shows on the skin in a bandlike formation and is often characterized by depressions or pits into the skin (Takehara and Sato, 2004).
Figure 1. Morphea Note the discolored plaques (courtesy of dermnet.com Scleroderma).
The second form of scleroderma, systemic sclerosis, has two subgroups. The first, limited sclerosis, typically only involving the fingertips or possibly the forearms, face, and neck. Diffuse systemic sclerosis is the classification used when the arms and trunk are also affected. The limited form of systemic sclerosis is less likely to result in life-threatening complications (Falanga, 1997). CREST syndrome is a commonly used term to describe those affected by the limited form of systemic sclerosis. CREST is an acronym for calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, telangiectasias. Not all symptoms must be present in order to classify a particular case as being CREST syndrome (Jimenez et al., 2006).
Figure 2. Pits on fingertips (courtesy of dermnet.com Scleroderma)
Scleroderma is a complicated term in that not all patients display the same effects to the same extent. In less serious cases, it is often only the skin that shows any sign of disease. In many cases, however, multiple organ systems within the body can be affected. The following are some of the possible symptoms in a patient with systemic sclerosis.
Vascular system: Most patients with scleroderma experience Raynaud’s phenomenon, a condition in which the skin changes colors from white to blue to red due to a decreased ability of the blood vessels to supply skin tissue with oxygen. Other characteristics include pain or tingling especially in the extremities (Jimenez et al., 2006). Raynaud’s phenomenon is often the first sign of systemic sclerosis (Condemi, 1992). It has been reported that 70% of systemic sclerosis patients already have Raynaud’s phenomenon once other symptoms arise, and 95% of the patients develop it over the course of their disease. The symptoms of Raynaud’s phenomenon could be present for months or even years before other symptoms of scleroderma are detected (Jimenez et al., 2006).
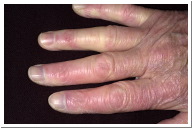
Figure 3. Raynaud's phenomenon (courtesy of dermnet.com Scleroderma)
Skin: As was mentioned earlier, the skin becomes thick and hard and these symptoms may also be accompanied by severe itching. Skin pigment changes in some areas is common in scleroderma, making the skin darker or lighter as a result of either hyperpigmentation or hypopigmentation, respectively. Telangiectasias can be clearly seen particularly on the face and the neck, which is the presence of enlarged blood vessels right under the skin (Jimenez et al., 2006).

Figure 4. CREST Note telangiectasias (courtesy of dermnet.com Scleroderma).
Other organ systems: Problems with the gastrointestinal system often result in complications with the esophagus, constipation, and diarrhea. Chest pain, difficulty breathing, or a dry cough can occur when the respiratory system is involved. In more serious cases pulmonary artery hypertension can arise. The musculoskeletal system is often affected, and as a result muscles become weak and tender. Patients often experience pain in their joints. Serious cases often arise once the cardiovascular and renal systems become negatively affected by systemic sclerosis. Renal crisis and hypertension can cause life-threatening complications (Jimenez et al., 2006).
Scleroderma and the immune system
Although the exact cause of scleroderma has yet to be determined, it has been classified as an autoimmune disorder. This is due mainly to the high presence of autoantibodies found in patients with both localized and systemic sclerosis. The higher presence of molecules associated with immune responses such as cytokines, cell adhesion molecules and cell surface antigens also points to autoimmunity as having a major role in the occurrences of scleroderma (Takehara and Sato, 2004).
Research suggests that the autoimmunity associated with localized scleroderma is different from that of systemic sclerosis. Takehara and Sato (2004) summarized previous studies dealing with the autoimmunity of localized scleroderma, looking in particular at antinuclear antibodies, cytokine and soluble receptors, and cell adhesion and surface molecules. They found that while both localized and systemic sclerosis involve the presence of autoantibodies, the particular autoantibodies observed in each case are different. A study by Sato et al. (2004) concentrated on autoimmunity associated with localized scleroderma, finding a very high prevalence of antihistone antibodies in patients with this disorder. They found that in all three types of localized scleroderma (morphea, generalized morphea, and linear scleroderma), there is a high prevalence of these autoantibodies. The particular histones H1, H2A and H2B seem to be the more accessible histones to which the antinucleosome antibodies are able to bind and begin an immune attack.
Studies done specifically on the autoantibodies present in patients with systemic sclerosis point to anticentromere antibodies as the main source of autoimmunity rather than antihistone antibodies. Other antibodies in this more serious form of sclerosis include anti-topomoisomerase antibodies (Takehara and Sato, 2004).
Abnormal B and T cell activation is thought to be the main source of the production of the autoantibodies associated with scleroderma (Takehara and Sato, 2004). In a study conducted by Boin et al. (2005) a high prevalence of CD8 T cells against autoantigens were found in patients with systemic scleroderma. These cytotoxic T cells were observed to be highly specific for topomoisomerase-1, which is a molecule responsible for breaking and rejoining one side of DNA and is therefore important during DNA replication, nucleoside assembly and DNA transcription (Takehara and Sato, 2004). Boin et al. found these CD T cells specific for topomoisomerase-1 in 30-60% of their subjects and found a high correlation to a phenotype involving diffuse systemic sclerosis and interstitial lung involvement. The researchers note that their results point to T cell dependent autoimmunity, although the exact effector pathways of the immune system that result in the phenotype described above are unknown.
Besides autoantibodies, scleroderma patients exhibit other obvious markers of immune system activation, as well as immune abnormalities. Studies have confirmed the presence of increased amounts of certain cytokines that play a role in immune responses, giving further support to the classification of scleroderma as an autoimmune disease. For example, increased levels of interleukin (IL)-2 have been found in scleroderma patients (Falanga, 1997). IL-2 is a growth factor produced by activated T cells, which act directly on the T cell, inducing the activated T cell to proliferate and differentiate (Janeway et al., 2005). The abnormalities found in patients with scleroderma include an infiltration of T cells, and a correlation has been found between the progression of the disease and the level of T cell infiltration in the patient’s body (Falanga, 1997).
The manner in which these immune abnormalities result in the various scleroderma phenotypes is unknown. It is thought, however, that the major physiological change involving the depositing of excess collagen on the skin and internal organs may have to do with the elevated cytokine levels in the body. The particular cytokines that have been studied and possibly correlated with the disease are ones such as IL-1, IL-4, and transforming growth factor (TGF)-β. TGF- β plays an important role in the deposition of collagen so is being vigorously studied. Another possibility is that the interferons involved with collagen synthesis regulation are being inhibited. Other studies suggest that because TGF- β function increases when little oxygen is present and the skin of scleroderma patients has very little access to oxygen, there is a correlation between TGF- β function and the increase in collagen synthesis and fibroblast formation (Falanga, 1997).
Janeway et al. (2005) discusses a positive feedback loop as a result of inflammation in many autoimmune diseases. Inflammation results in the recruitment of macrophages and neutrophils, which in turn release cytokines and chemokines which further stimulate the immune response. When this immune response is triggered by self, as is the case in autoimmune diseases, the ever-present antigen continues to result in inflammation and the further release of cytokines and chemokines. Two main events have been undeniably associated with scleroderma, those being endothelial cell damage and the depositing of excess collagen in areas of the body (Falanga, 1997). Again, the primary cause of these events is unknown, but there has been some success in identifying the role of the immune system in furthering the effects of the disorder. One study conducted by Gabriele et al. (2000) looked at the role of oxidative stress in the continuation of systemic sclerosis. Researchers recognized the damage done to endothelial cells as a result of free radicals released in the body as part of the effects of Raynaud’s phenomenon. This damage leads to vessel narrowing and a decreased amount of oxygen able to reach the body’s tissues, which in turn leads to even more free radicals and thickening of organ tissue. The possitive feedback cycle discussed in Janeway et al., then, continues the destruction of endothelial tissue, which is one of the main characteristics of scleroderma.
Other immune abnormalities observed in scleroderma patients include a very high CD4:CD8 T cell ratio (Dau and Callahan, 1994). The importance of this fact was illustrated in a study by Dau and Callahan, during which patients who received plasmapheresis treatments and immunosuppressive drugs showed vast improvement over time, and returned to a normal CD4:CD8 T cell ratio. This same study also found that after improved condition due to the treatments, the number of T cells believed to play a role in the suppression of T cell activation increased, suggesting that their function is inhibited in people with systemic scleroderma.
Again, the exact effector mechanisms that lead to the varying scleroderma phenotypes are unknown. There remains much research to be done before the complicated pathways and interactions can be fully understood.
Treatment
The exact cause of scleroderma makes treatment a difficult task. Again, there is huge variability among patients exhibiting scleroderma and the treatments they use vary as well. There is no known cure for scleroderma; therefore the focus is on alleviating symptoms and avoiding further damage.
One study examined the changes observed in systemic scleroderma patients over the course of treatment with plasmapheresis, cyclophosphamide and prednisone. Prednisone is a commonly used anti-inflammatory corticosteroid drug. Corticosteroids are lipid-soluble and act by binding to their receptor after diffusing across the plasma membrane (see Figure 5). Once bound to the receptor, the complex enters the nucleus and binds to areas of DNA sequences that promote transcription and in this way can regulate genes (Janeway et al., 2005). As a result of treatments in the study, there was a sharp increase in the amount of CD8 T cells circulating in the patients. This normalized the CD4:CD8 ratio in the patients and presumably played a role in their improvement. There was also a decline in the amount of B cells, presumably resulting in a decline in the number of autoantibodies being produced (Dau and Callahan, 1994).
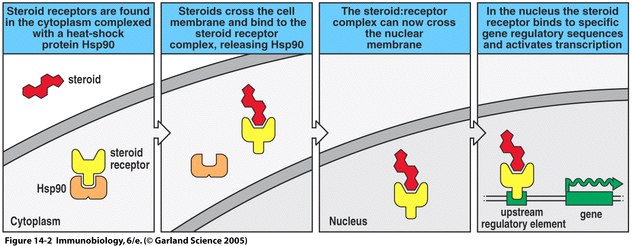
Figure 5. Mechanism of prednisone function. Prednisone, a corticosteroid, binds to its receptor, ultimately allowing regulation of gene transcription (Janeway et al., 2005).
Gabriele et al. suggest the use of antioxidant therapies as a way of avoiding further endothelial damage in systemic sclerosis occurrences. The positive feedback was discussed above (see scleroderma and the immune system), and researchers suggest that if the oxygen free radicals could be diminished from promoting further endothelial damaging, the inflammation could be lessened and the immune response inhibited (Gabriele et al., 2000).
Other ways of dealing with scleroderma symptoms involve easier steps such as keeping the extremeties warm in cases that involve Raynaud’s phenomenon, keeping the skin moist. Certain drugs also seem to alleviate symptoms. Interferon (IFN)-γ seems to slow the production of collagen, which could decrease the hardening effects of the disease on the skin. Calcium channel blockers also help in alleviating Raynaud’s phenomenon. Although these discoveries are promising, not much seems to be known regarding the exact mechanisms of how they work to avoid furthering the disease (Herrick, 1998).
Researchers hope to make progress in understanding the mechanisms involved with the progression of scleroderma, particularly the more dangerous systemic scleroderma in hope to finding a way of avoiding or at least more effectively treating the symptoms. One study looked at using a mouse model to further study systemic scleroderma, particularly the autoantibodies associated with the disease. The tight skin-2 mouse was observed to have excess collagen deposition in its skin, as well as anti-topomoisomerase-1, anti-nucleolus, and anti-centromere antibodies. The researchers hope to use this mouse to study autoimmunity related to not only systemic sclerosis, but other related connective tissue diseases (Gentiletti et al., 2005).
References
Boin F, Wigley FM, Schneck JP, Oelke M, Rosen A, 2005. Evaluation of topoisomerase-1-specific CD8+ T-cell response in systemic sclerosis. Human Immunology: Patient-Based Research 1062:137-145.
Condemi JJ, 1992. Progressive systemic sclerosis (scleroderma). American Medical Association Journal 268:2889-2890.
Dau PC, Callahan JP, 1994. Immune modulation during treatment of systemic sclerosis with plasmapheresis and immunosuppressive drugs. Clinical Immunology and Immunopathology 70:159-165.
Falanga V, 1997. Scleroderma in Skin Immune System (SIS). Ed. Jan Bos. CRC Press LLC:461-470.
Gentiletti J, McCloskey LJ, Artlett CM, Peters J, Jimenez SA, Christner PJ, 2005. Demonstration of autoimmunity in the tight skin-2 mouse: A model for scleroderma. Journal of Immunology 175:2418-2426.
Herrick AL, 1998. Advances in treatment of systemic sclerosis. Lancet 352:1874-1875.
Janeway, C.A., Travers, P., Walport, M., Schlomchik, M. Immunobiology 6th Ed: The Immune System in Health and Disease. New York: Garland Publishing, 2005.
Jimenez S, Koenig AS, Cronin PM. 2006 Jan 27. Scleroderma. <http://www.emedicine.com/MED/topic2076.htm> Accessed 2006 April 19.
Sato S, Kodera M, Hasegawa M, Fujimoto M, Takehara K, 2004. Antinucleosome antibody is a major autoantibody in localized scleroderma. British Journal of Dermatology 151:1182-1188.
Simonini G, Pignone A, Generini S, Falcini F, Matucci CM, 2000. Emerging potentials for an antioxidant therapy as a new approach to the treatment of systemic sclerosis. Toxicology 155:1-15.
Takehara K, Sato S, 2004. Localized scleroderma is an autoimmune disorder. Rheumatology 44:274-279.
Questions or comments? jeking@davidson.edu
Return to immunology home page

