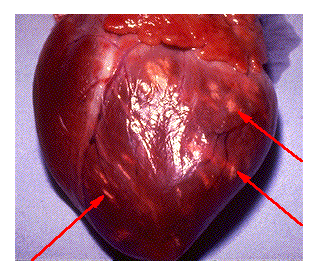
This web page was produced as an assignment for an undergraduate
course at Davidson College*
STAGES OF MYOCARDITIS
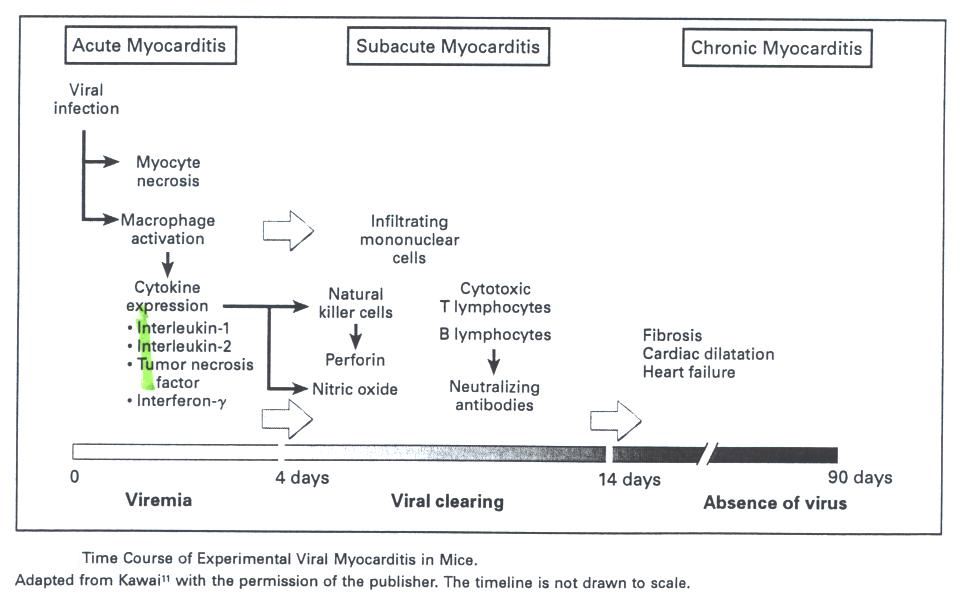
Fig. 1 Timeline of myocarditis. Each stage of the myocarditis has
specific key players that are involved in the activation process.
This figure was
borrowed from Medical Progress Review Article Nov 9, 2000.
NATURE OF DISEASE
Myocarditis is clinically defined as
inflammation of the heart muscle. There
is a large variety of infections, systemic diseases, drugs, and toxins that
associate with the development of this disease.
Viruses, bacteria, protozoa, and even worms have been implicated as
infectious agents (Feldman and McNamara, 2000).
The diagnosis of myocarditis can be made by endomyocardial biopsy and the
most frequent cardiotropic viruses detected include Parvo B19, measles, chicken
pox, enteroviruses, adenoviruses, cytomegalovirus, Epstein Barr virus, and
influenza virus (Maisch et al., 2003). Viral
diseases are more commonly associated with myocarditis in immunocompetent hosts
such as human immunodeficiency virus type 1 (HIV-1) and hepatitis C (Hep C).
Recent researches showed that HIV-1 and HIV-1 RNA has been detected in
heart tissue from patients with acquired immunodeficiency syndrome (Jacqueline
and Blanchfield, 2002). Dilated
cardiomyopathy was evident that in 80 percent of a large group of asymptomatic
HIV-postive patients, 83 percent of who had myocarditis and 6 percent of whom
had detectable HIV myocardium. However,
there are some studies that fail to show HIV proviral DNA in myocardial samples
obtained from HIV-infected children. Hence
it is unclear whether it is HIV itself or the appearance of secondary viruses in
an immunocompromised host that accounts for the high incidence of myocarditis in
HIV-positive patients. Another
viral disease that is involved in the development of dilated myocardiopathy is
Hep C. It was found that Hep C
virus replicate in patients myocardium (Doroshenko, 2002).
The most common myocarditis resulted from bacteria is the Chagas’
disease, an inflammatory disease caused by the parasitic protozoan Trypanosoma
cruzi. In addition, drugs can
also cause myocardial inflammation by direct toxic effect on the myocyte or
through immune-mediated mechanisms. An
example of a drug-induced toxicity is cocaine, which causes cardiac dysfunction
by its vasoconstrictor properties. Patients
with drug-induced allergic myocarditis tend to have eosinophilia or an
eosinophilic infiltrate in the myocardium (Feldman and McNamara, 2000).
IMMUNE RESPONSE
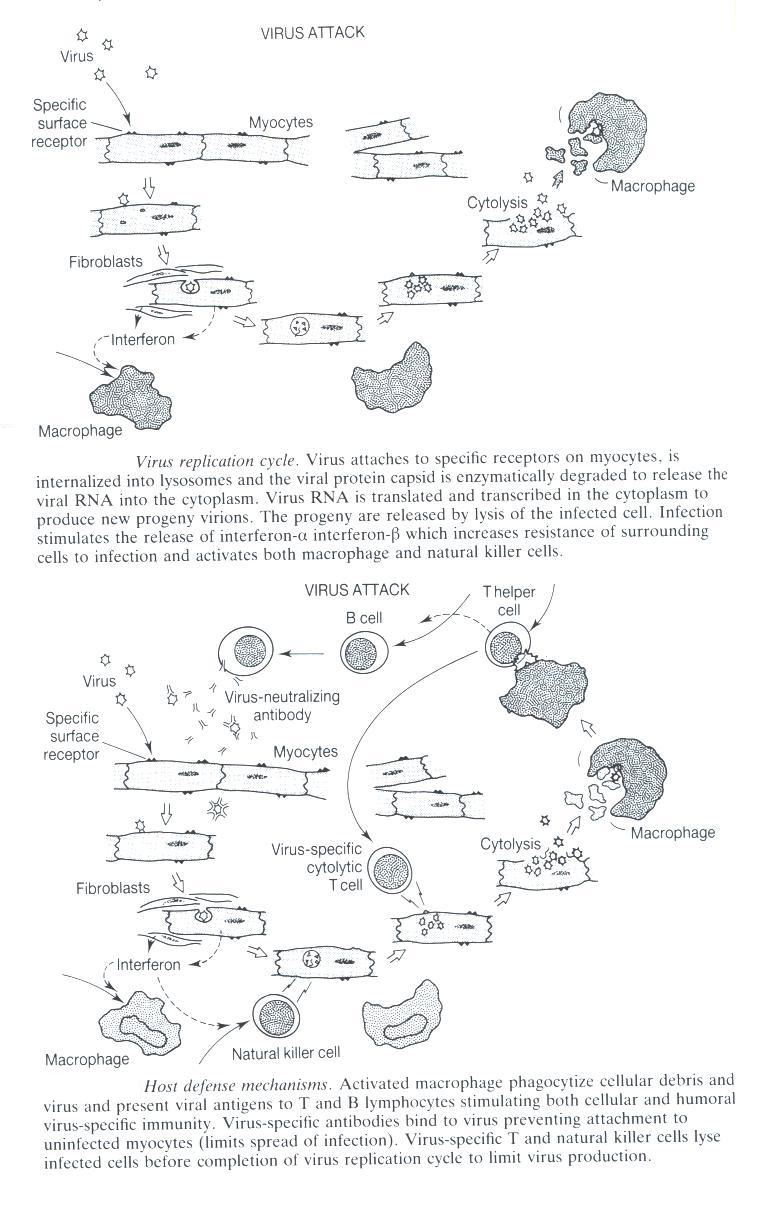
Fig. 2 Replication of virus followed by the host defense mechanisms.
This figure was borrowed from Viral Infection of the Heart by J.E. Banatvala p.
84
An example that shows the mechanisms in
which the immune system responds can be illustrated by the coxsackievirus B.
The first stage includes RNA viruses are taken into cells by
receptor-mediated endocytosis and directly translated inside the cells to
produce viral protein. The virus
then replicates in the cytoplasm of the myocytes, but eventually it can also be
released into the interstitium, where it undergoes by phagocytosis by
macrophages (Feldman and McNamara, 2000). The
second stage of viral infection is characterized by infiltration by inflammatory
cells, including natural killer cells and macrophages with the subsequent
expression of proinflammatory cytokines. Recent
studies demonstrate that CD1d expression increases early after the infection and
that CD1d is essential for pathogenicity of CVB3-induced myocarditis (Huber et
al., 2003). ERK-1/2 is an
extracellular signal-regulated kinase which influences the p56(Lck) and in turns
activate T cells. The enhancement
of ERK-1/2 activation, therefore, induces myocarditis to those who are
susceptible to coxsackievirus (Opavsky et al., 2002).
Moreover, the VP1 protein in Coxsackievirus B group potentially
functioned as a predominant antigen inducing detectable IgM antibody following
CVB infection (Zhang et al., 2001). XJEK
granules are efficacious in inhibiting CVB3m protecting and curing virus
myocarditis. The granules are also
good for inflammation, anti-myocardial ischemia, anti-arrhythmia, and increase
the serum IgG level (Wang et al., 2000). Interestingly,
Coxsackievirus group B type 3 (CVB3) have different effects based on genders.
CVB3 induces myocarditis in male but produces little cardiac injury in
females. The males develop
cytolytic T lymphocytes (CTL) reactive to heart antigens which primarily cause
the inflammation and cardiac injury. The
infected females lack this CTL response because they rapidly produce suppressor
cells inhibiting both cellular immunity and cardiac inflammation (Job et al.,
1986).
Fig. 3 CVB3 A
schematic representation of a feasible structure for the human coxsackievirus
which is depicted that is derived from the crystal structure of the Ig V-like
domain. Image was taken from http://wehih.wehi.edu.au/scop/data/scop.b.html
The macrophage activation results from
the release of viral particles into the interstitium and the release of
interferon-y by natural killer cells and other activated white cells.
After activation by interleukin-2, the natural killer cells protect
against viral invasion by eliminating virally infected cells, and thus
inhibiting virus replication. Not
only natural killer cells release perforin and granzymes, they can exacerbate
disease by injuring the cardiomyocytes. Hence,
natural killer cells only interact with virus-infected myocytes, sparing the
uninfected cells (Feldman and McNamara, 2000).
Cell-mediated immunity also has an
important role in viral clearing. Within
seven days of infection, antigen-specific T cells infiltrate the mouse
myocardium. Those circulating T
cells, which express the alpha and beta chains of the T cell receptor and either
CD4 or CD8 coreceptor molecules, include T helper cells and cytotoxic T
lymphocytes. The cytotoxic T cells
then recognize the degraded fragments of viral proteins that are presented by
the major-histocompatibility-complex class I (MHC I) antigens on the surface of
the myocyte membrane. Cytokines
such as interferon-y (IFN-y) induce the up-regulation of the MHC antigens on the
surface of the myocytes. It was
found that type I IFNs act as a natural adjuvant for the immune response against
myocarditis. Type I IFN DNA
coimmunisation may provide increased efficacy for viral vaccines and
subsequently modulate post-viral chronic inflammatory disorders (Feldman and
McNamara, 2000).
In order to become fully activated, T
cells must also receive a second signal from the costimulatory molecules such as
B7 on the antigen presenting cells. When
there are appropriate cofactors and antigens, cytotoxic T lymphocytes become
activated and are able to lyse virus-infected cardiocytes (Feldman and McNamara,
2000). Moreover, the cell-to-cell
contact allow effective lysis and is mediated by the up-regulation of
intercellular adhesion molecules (ICAM-1) on the surface of the infected myocyte,
which is induced by tumor necrosis factor alpha (TNF alpha) and INF-y (Cooper,
2003). These molecules help to further
mobilize immunocytes to the injury area to remove the damaged or
toxin/virus-contaminated cells. The ICAM-1 up-regulation can further lead
to the coupling of T cells with expressed LFA-1 ligand, leading to killing of
the infected host cell by natural killer cells via perforin mechanism (Schultheiss
and Schwimmbeck, 1997). The development of neutralizing antiviral
antibodies also helps the clearing of the viral infection.
The following diagram indicates the mechanisms of humoral and cellular immunity:
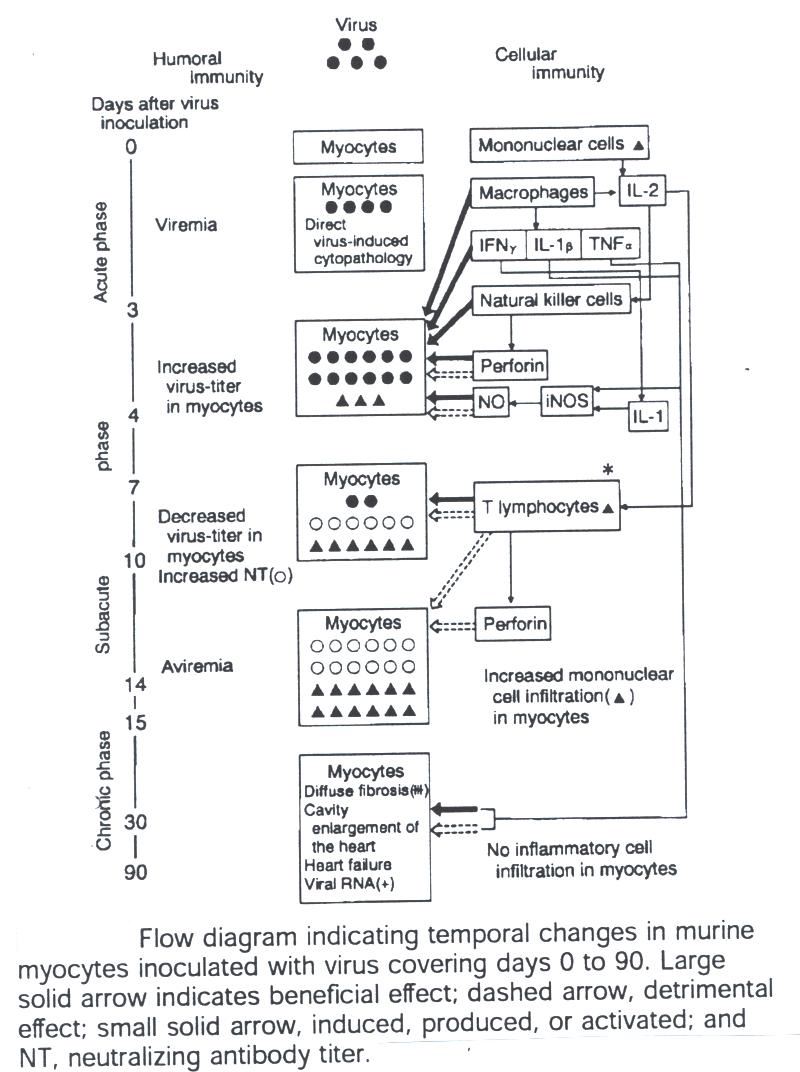
Fig. 4 Mechanisms
of humoral and cellular immunity. This figure was borrowed from
Medical Progress Review Article Nov 9, 2000.
AUTOIMMUNE RESPONSE
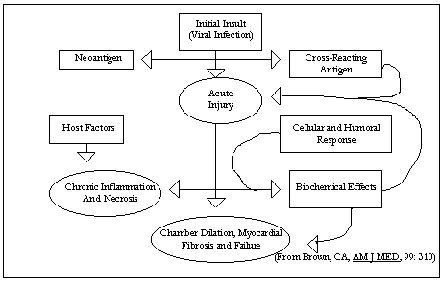
Fig. 5 presents a paradigm for the
development of cardiomyopathy. hsc.virginia.edu/.../internal/conf/chiefs/myocarditis.htm
by Dr. Scott Robertson
(
There was no email address available to request for permission regarding the
image.)
The ultimate function of the immune
response is to clear virus and allow healing in many instances of myocardial
infection. However, inefficient
viral clearing or overaggressive immunologic activation can be observed as well.
For example, in some strains of mice, normal host defense mechanisms are
inadequate, and persistent viral replication occurs in the myocytes, resulting
in chronic viral disease with cardiac dilation and failure.
The expression of toll-like receptor 4 increases the enteroviral
replication in human myocarditis (Satoh et al., 2003).
The persistent activation of infiltrating T cells during viral infection
can cause long-term tissue destruction, leading to dilated cardiomyopathy.
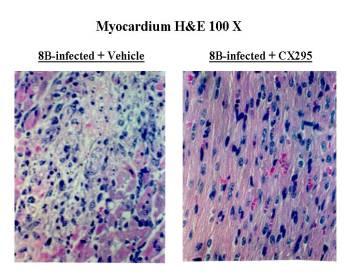
Fig.
6 Infection of neonatal mice with reovirus strain 8B produces myocarditis in
neonatal mice due to a direct viral injury of myocytes.
The myocardial injury is due to apoptosis. Apoptosis was then
verified by TUNEL staining co-localizing to areas of viral antigen, as well as
by demonstration of characteristic oligonucleotide laddering of nuclear DNA. The
data shows that calpain inhibitors block retrovirus-induced apoptosis in vitro.
Targeting of apoptotic signaling pathways may serve as a novel antiviral
strategy.
www.uchsc.edu/.../robertadebiasi/
invivomyocarditis.htm
(permission to use image was granted on 4-15-03 by Dr. Roberta DeBiasi)
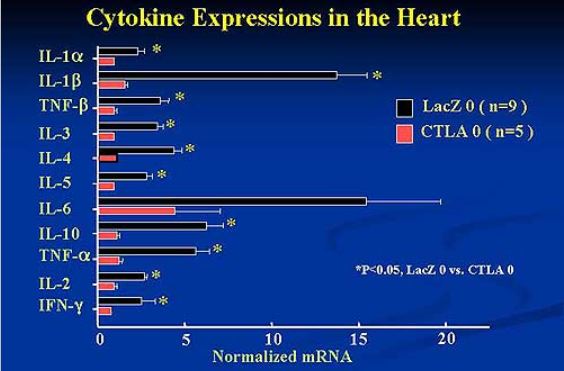
Fig.
7 Cytokine Expressions in the Heart. AdexCTLA-4IgG suppressed cytokine
activation and the onset and progression
of experimental autoimmune myocarditis in the rat model.
www.j-circ.or.jp/.../sessions/ reports/66th-ss/ps06-z4.htm
(requested
permission on 4-15-03)
The proinflammatory cytokines also have important roles in
the development of chronic inflammatory disease.
Tumor necrosis factor activates endothelial cells, recruits inflammatory
cells, enhances the production of inflammatory cytokines, and has significant
negative inotropic effects. Interferons
have a critical role in attenuating viral replication when administered
endogenously (Feldman and McNamara, 2000).
In addition, cytokines can activate inducible nitric oxide synthase in
cardiac myocytes, which can simultaneously give cardiacprotection as well as the
development of myocarditis (Ding, 2002). Nitric
oxide is a free radical gas that plays paracrine/autocrine and intracrine roles
in maintaining physiological cardiovascular performance.
In the coronary circulation, NO mediates endothelium-dependent
vasodilator responses to shear stress and agonist-induced responses to
neurohumoral stimulation. In the
heart, NO modulates myocardial relaxation, beta-adrenergic responses,
mitochondrial respiration and substrate metabolism and excitation-contraction
coupling (Champion and Hare, 2001). The
beneficial and deleterious roles of nitric oxide are still debatable. Moreover,
the cellular effector mechanisms such as natural killer cells activity was
markedly decreased in the acute state in target cell of patients with
perimyocarditis. In contrast, in myocarditis target cell specific
non-major histocompatibility complex (MHC) restricted lysis against living adult
allogenic rat myocytes is slightly enhanced (Banatvala, 1993).
There are studies that show humoral
immunity is also essential in the development of postinfectious myocarditis.
The experiments showed that a T-cell-dependent myocarditis could be
demonstrated in mice after immunization with cardiac C protein or streptococcal
M protein peptide, adoptive transfer of myosin-reactive cells or plenocytes
after myocardial infarction, or transplantation of a normal heart into a
virus-treated host after documentation of viral clearance.
The researchers suggested that there is an autoimmune myocarditis with
the evidence of cross-reactive epitopes between cardiac myosin and infectious
agents (Kishimoto et al., 2001). Upon
secondary exposure of enteroviruses, T cell-mediated immune responses to a
conserved antigenic epitope (Kishimoto et al., 2001).
Not only there is a relation between virus and the subsequent development
of cardiomyopathy, there are other factors such as physical-activitylevel, sex,
age, and genetic background which determine the virulence of the infection.
Patients with myocarditis normally have
an imbalance between helper and cytotoxic T cells; an inappropriate expression
of the MHC on cardiac tissues; and circulating organ-specific autoantibodies in
the serum. The cytotoxic activity
against healthy cardiomyocytes was myocyte-specific, induced by CD8 lymphocytes
and MHC restricted. Cytotoxic T
lymphocytes are activated following myocardial infarction and can recognize and
kill healthy myocytes in vitro (Varda-Bloom et al., 2000).
In patients with diated cardiomyopathy, autoantibodies have been
identified that react with heart mitochondria, the adenine nucleotide
translocator, the muscarinic receptor, myosin heavy chain, or laminin.

Fig. 8 Mechanisms that lead to autoimmune response. This figure was
borrowed from Viral Infections of The Heart by J.E. Banatvala, p. 85
DIAGNOSIS
Patients with myocarditis disease
normally have flulike syndrome accompanied by fever, arthralgias, and malaise.
Clinicians can also perform laboratory tests to find out if patients have
an elevated sedimentation rate, eosinophilia, an elevation in the cardiac
fraction of creatine kinase, cardiac troponin T or troponin I.
Other diagnostic tests include autoimmune serum markers or the induction
of the MHC and intercellular adhesion molecules on cardiac myocytes to identify
patients with autoimmune myocarditis.
To further confirm the diagnosis, usually the physician would obtain a
cardiac catherization (Feldman and McNamara, 2000).
Researchers believe that complement
plays a critical role in the development of autoimmune myocarditis and that it
acts through complement receptor type 1 (CR1) and type 2 (CR2).
There is a subset of CD44 and CD62L T cells that expresses CR1 and CR2,
which suggest that both receptors are involved in the B and T cell activation, T
cell proliferation, and cytokine production.
Since the activated complement is the key product of the innate immune
response, it modulates the induction of an autoimmune disease (Kaya et al.,
2001).
Activation of dendritic cells is another
important component of myocarditis. Interleukin
1 receptor 1 (IL-1R1) triggering is
required for efficient activation of dendritic cells, which is in turn a
prerequisite for induction of autoreactive CD4+ T cells and autoimmunity
(Eriksson et al., 2003). CD4+ T
cells induce local eosinotaxis, mediated by IL-5, and participate in
myocardiocyte injury via Eo induction (Hirawasa et al., 2003).
IL-6 is required for the expansion of autoimmune CD4+ T cells and the
pathogenesis of autoimmune myocarditis by upregulate complement C3 (Eriksson, et
al., 2003). CD83+ dendritic cells
(CDs) are involved in the inflammatory process and triggered by an imbalance of
DCs and their failure to confer tolerance to self-antigen (Schoppet et al.,
2003). Cardiac antigen-specific
CD8+ T cells are involved in the autoimmune component of human myocarditis and
that IL-12 is required for the differentiation of pathogenic CD8+ T cell
effectors (Grabie et al., 2003).
TREATMENT
Treatments of myocarditis include
antibiotics to fight of the infection if the cause is bacterial infection.
However, if the case is severe, the only option that allows complete
recovery is heart transplantation (Long and Blanchfield, 2002).
Drugs that reduce the basal levels of cytokines are amlodipine,
pentoxifylline, and beta-blockers. There
are also drugs that help reduce endotoxin-induced cytokine gene expression:
ouabain, amiodarone, adenosine, angiotensin converting enzyme inhibitors (e.g.
captopril, enalapril, and lisinopril), angiotensin II-receptor blockers (Godsel
et al., 2003). Direct blockade of
the deleterious actions of elevated plasma levels of cytokines recently became
possible through intravenous infusion of a soluble TNF-alpha receptor fusion
protein, which resulted in an increase in exercise tolerance and left ventricle
performance (Paulus, 2000). Immunosuppressive
therapy is not recommended in patients with infectious or postinfectious
myocarditis. However,
immunosuppression is essential for patients with cardiac dysfunction due to a
systemic autoimmune disease and it can be achieved by the help of biventricular
mechanical assist (BVS 5000) (Marelli et al., 2003).
Successful treatment of enterovirus-induced myocarditis with
interferon-alpha was proven to be a reliable therapy (Daliento et al., 2003).
Embryonic stem cells significantly increase the survival of viral
myocarditis mice and also decrease the necrosis and infiltration of inflammatory
cells (Wang et al., 2002). In
addition, the suppressor of cytokine signaling-1 (SOCS1) is a novel therapeutic
target for enterovirus-induced cardiac injury by inhibiting the signaling of
Janus Kinase (JAK) and signal transducers and activators of transcription (STAT)
(Yasukawa et al., 2003). An
antiviral agent can reduce the number of infected cells and virus identified in
human myocardial fibroblasts. An
alternative approach to the treatment of viral myocarditis has been the
development of virus-specific vaccines. Attenuated
vaccines have successfully prevented the development of myocarditis after viral
challenge in mice, pigs, and elephants. The
usefulness of vaccines in humans remains unclear (Feldman and McNamara, 2000).
PREVENTION
Even though myocarditis is an unpredictable disease, the
following steps can help prevent its onset:
*take extra measures to avoid infections, and obtain
appropriate treatment for infections.
*limit alcohol consumption to no more than one or two
drinks a day, if any.
*maintain current immunizations against diphtheria,
tetanus, measles, rubella, and polio.
*avoid anything that may cause the abnormal heart to work
too hard, including salt and vigorous exercise (Long and Blanchfield, 2002).
REFERENCES
Banatvala, J.E. 1993. Viral Infections of The
Heart. Boston, MA: Hodder and Stoughton, pp.84-161.
Champion, H.C., and J.M. Hare.
2001. Emerging therapeutic
targets in nitric oxide-dependent cardiac disease.
Expert Opin Ther Targets 5(5):547-556.
Cooper, L.T. 2003. Myocarditis From Bench to
Bedside. Totowa, NJ: Humana Press, pp.101-2.
Cull, V.S., S. Broomfield, E.J. Bartlett, N.L. Brekalo, and
C.M. James. 2002.
Coimmunisation with type I IFN genes enhances protective immunity against
cytomegalovirus and myocarditis in gB DNA-vaccinated mice.
Gene Ther 9(20):1369-78.
Daliento, L., F. Calabresse, F. Tona, A.L. Caforio, G.
Tarsia, A. Angelini, and G. Thiene. 2003.
Successful treatment of enterovirus-induced myocarditis with
interferon-alpha. J Heart Lung
Transplant 22(2):214-7.
Ding, G.F. 2002.
Involvement of immune system in the pathogenesis of viral myocarditis.
Sheng Li Ke Xue Jin Zhan 33(1):30-7.
Doroshenko, B.H. 2002.
Hemostasis status in patients with acute viral myocarditis.
Lik Sprava 5-6:22-3.
Eriksson, U., M.O. Kurrer, N. Schmitz, S.C. Marsch, A.
Fontana, H.P. Eugster, and M. Kopf. 2003.
Interleukin-6-deficient mice resist development of autoimmune myocarditis
associated with impaired upregulation of complement C3.
Circulation 107(2):320-5.
Eriksson, U., M.O. Kurrer, I. Sonderegger, G. Iezzi, A.
Tafuri, L. Hunziker, S. Suzuki, K. Bachmaier, R.M. Bingisser, J.M. Penninger,
and M. Kopf. 2003.
Activation of dendritic cells through the interleukin 1 receptor is
critical for the induction of autoimmune myocarditis.
J Exp Med 197(3):323-31.
Feldman, A.M., and D. McNamara.
2000. Myocarditis.
The New England Journal of Med 343(19):1388-98.
Godsel, L.M., J.S. Leon, and D.M. Engman.
2003. Angiotensin Converting
Enzyme Inhibitors and Angiotensin II Receptor Antagonists in Experimental
Myocarditis. Curr Pharm Des
9(9):723-35.
Grabie, N., M.W. Delfs, J.R. Westrich, V.A. Love, G.
Stavrakis, F. Ahmad, C.E. Seidman, J.G. Seidman, and A.H. Lichtman.
2003. IL-12 is required for
differentiation of pathogenic CD8+ T cell effectors that cause myocarditis.
J Clin Invest 111(5):671-80.
Hirasawa, M., H. Deguchi, A. Ukimura, and Y. Kitaura.
2003. Immunologic
interaction between infiltrating eosinophils and T lymphocytes in murine
spontaneous eosinophilic myocarditis. Int
Arch Allergy Immunol 130)1):73-81.
Huber, S., D. Sartini, and M. Exley.
2003. Role of CD1d in
coxsackievirus b3-induced myocarditis. J
Immunol 170(6):3147-53.
Job, L.P., D.C. Lyden, and S.A. Huber.
1986. Demonstration of
suppressor cells in coxsackievirus group B, type 3 infected female Balb/c mice
which prevent myocarditis. Cell
Immunol 89)1):104-13.
Kaya, Z., M. Afanasyeva, Y. Wang, K.M. Dohmen, J.
Schlichting, T. Tretter, D. Fairweather, V.M. Holers, and N.R. Rose. 2001.
Contribution of the innate immune system to autoimmune myocarditis: a
role for complement. Nat Immunol
2(8):739-45.
Kishimoto, C., Y. Hiraoka, and H. Takada.
2001. T cell-mediated immune
response enhances the severity of myocarditis in secondary cardiotropic virus
infection in mice. Basic Res
Cardiol 96(5):439-45.
Long, J.L., and D.S. Blanchfield.
2002. The Gale Encyclopedia
of Medicine. Farmington Hills, MI:
Gale Group, pp. 2288-91.
Maisch, B., A.D. Ristic, I. Portig, and S. Pankuweit.
2003. Human viral
cardiomyopathy. Front Biosci
8:S39-67.
Marellin, D., R. Kermani, J. Bresson, M.C. Fishbein, M.
Hamilton, J. Moriguchi, G.C. Fonarow, B. Cohen, J. Kobashigawa, and H. Laks.
2003. Tex Heart Inst J
30(1)50-6.
Opavsky, M.A., T. Martino, M. Rabinovitch, J. Penninger, C.
Richardson, M. Petric, C. Trinidad, L. Butcher, J. Chan, and P.P. Liu.
2002. Enhanced ERK-1/2
activation in mice susceptible to coxsackievirus-induced myocarditis.
J Clin Invest 109(12):1561-9.
Paulus, W.J. 2000.
Cytokines and heart failure. Heart
Fail Monit 1(2):50-6.
Satoh, M., M. Nakamura, T. Akatsu, J. Iwasaka, Y. Shimoda,
I. Segawa, and K. Hiramori. 2003.
The expression of toll-like receptor 4 associated with enteroviral
replication in human Myocarditis. Clin
Sci (Lond) [epub ahead of print].
Schoppet, M., S. Pankuweit, and B. Maisch.
2003. CD83+ dendritic cells
in inflammatory infiltrates of Churg-Strauss myocarditis.
Arch Pathol Lab Med 127(1):98-101.
Schultheiss, H.P., and P. Schwimmbeck. 1997.
The Role of Immune Mechanisms in Cardiovascular Disease. Berlin, Germany:
Springer, pp. 47-48.
Varda-Bloom, N., J. Leor, D.G. Ohad, Y. Hasin, M. Amar, R.
Fixler, A. Battler, M. Eldar, and D. Hasin.
2000. Cytotoxic T
lymphocytes are activated following myocardial infarction and can recognize and
kill healthy myocytes in vitro. J
Mol Cell Cardiol 32(12):2141-9.
Wang, J.F., Y. Yang, G. Wang, J. Min, M.F. Sullivan, P.
Ping, Y.F. Xiao, and J.P. Morgan. 2002.
Cell Transplant 11(8):753-8.
Yasukawa, H., T. Yajima, H. Duplain, M. Iwatate, M. Kido,
M. Hoshijima, M.D. Weitzman, T. Nakamura, S. Woodard, D. Xiong, A. Yoshimura,
K.R. Chien, and K.U. Knowlton. 2003.
The suppressor of cytokine signaling-1 (SOCS1) is a novel therapeutic
target for enterovirus-induced cardiac injury.
J Clin Invest 111(4):469-78.
Wang, Q.M., G.L. Chen, Y.J. Wang, H.S. Wang, M.H. Gao, and Y.Z. Gong.
2000. An experimental study
on inhibitor effect of xinjierkang granules on virus myocarditis.
Zhongguo Zhong Yao Za Zhi 25(5):293-6.
Zhang, T.,
G. Ma, and L. Ma. 2001.
Study on specificity of IgM antibody response in patients with coxsackie
virus B infection. Zhonghua Shi
Yan he Lin Chuang Bing Du Xue Za Zhi 15(1):66-8.
____________________________________________________________________________
Are
you intrigued by the effects of Zeta-Associated Protein 70?
ZAP-70
Human's
Syk tyrosine kinase is part of the SH2 domain family which includes ZAP-70.
Image
was taken from http://wehih.wehi.edu.au/scop/data/scop.b.html
Structure
and Function
Mutations
of ZAP-70 and Related Disorders
Other
related defects that cause SCID
ZAP-70 Related Treatments for Immunodeficiency Disorders
Alterations
of ZAP-70 Signaling
References
Zeta-associated protein or ZAP-70 is one of the principal targets of Lck in T
cells, which is essential in propagating the signal onward.
It is a non-src family protein kinase which associates with
phosphorylated CD3 zeta chain, and plays an important role in TCR-CD3 complex
signaling (Delves and Roitt 1998). ZAP-70
binds to the phosphorylated zeta chain ITAMs and is phosphorylated and activated
by Lck when the coreceptor binds to the MHC ligand (Janeway et al., 2001).
The expression of ZAP-70 in developing T cells promotes the development
of single positive from double positive thymocytes.
Current studies show that ZAP-70 tyrosine kinase is required for the
up-regulation of Fas ligand in activation-induced T cell apoptosis (Eischen et
al., 1997). An interesting research
discovered that T cells from a substantial proportion of elderly humans exhibit
significant reductions in the catalytic activity, but not expression of ZAP-70
when stimulated by ligation of the TCR/CD3 with cross-linked anti-CD3 monoclonal
antibody OKT3. Hence, age-related
impairments of ZAP-70 activation in anti-CD3 stimulated T cells are associated
with reduced tyrosine phosphorylations of zeta chains and autophosphorylations
of the PTKs p56lck/p59fyn (Whisler et al., 1999).
Structure and Function
ZAP-70 has two SH2 domains in its amino terminal halves and a carboxy-terminal
kinase domain. The crystal structure
of the tandem SH2 domains of human ZAP-70 in complex with a peptide derived from
the zeta-subunit of the T cell receptor reveals an unanticipated interaction between
the two domains. A coiled coil of
alpha-helices connects the two SH2 domains, producing an interface that constitutes
one of the tow critical phosphotyrosine binding sites (Hatada et al., 1995).
As each SH2 domain binds to one phosphotyrosine, ZAP-70 preferentially
binds to motifs with two phosphotyrosines spaced a precise distance apart.
ZAP-70 is thus recruited to the receptor complex upon full phosphorylation
of the ITAMs. Binding to the ITAM
peptide induces large movements between the two SH2 domains and the actual binding
sites. The conformation of the ITAM-free
protein is partly governed by a hydrophobic cluster between the linker region
and the C-terminal SH2 domain (Folmer et al., 2002).
The activation of ZAP-70 is mainly contributed by Lck, which in turn phosphorylates
LAT (Linker of Activation in T cells) and SLP-76 (Second Linker Protein) (Pelosi
et al., 1999). SLP-76 and LAT are
each critical for the expansion and differentiation of double-negative thymocytes
and that SLP-76 is essential for allelic exclusion at the TCR beta locus (Pivniouk
and Geha, 2000). Tec kinases then
activate PLC-y and guanine-nucleotide exchange factors activate Ras which propagates
the signal from the cell membrane into the nucleus to begin gene transcription.
Another function of ZAP-70 is that in the immune synapse, ZAP-70 controls
T cell polarization and recruitment of signaling proteins but not formation of
the synaptic pattern (Blanchard et al., 2002).
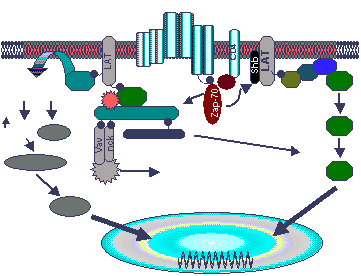
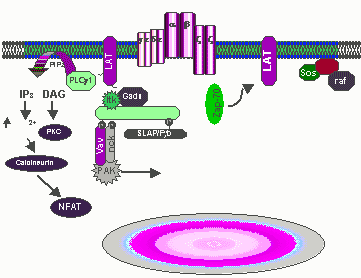
Fig. 1 Activated ZAP-70 phosphorylated SLP-76
and LAT initiating ras/MAPK and PLCgl signaling cascades. Tyrosine
phosphorylated LAT which is constitutively located in glycolipid enriched
membrane microdomains additionally recruits PLCgl1to the membrane, placing PLCg1
in close proximity with its substrate, PIP2. PIP2 is hydrolyzed IP3 and
DAG which leads to increases of cytosolic free calcium and activation of protein
kinase C. Tyrosine phosphorylated LAT binds to the Grb2 SH2 domain which
recruits Sos to the plasma membrane thus innitiating Ras/ERK activation.
Since Gads constitutively associates with SLP-76, theSLP-&6/Gads complex may
be recruited to LAT leading to ras/MAPK and PLCg1 activation. Tyrosine
phosphorylated SLP-76/Vav/Nck/Pak complex which may be important for the
regulation of cytoskeletal rearrangements. SLP-76 additionally binds to
the SH2 domain of Itk which subsequently phosphorylates and further activates
PLCg1. (Requested permission from Dr. Gary Koretzky, M.D., Ph.D. on
3/13/03)< http://www.med.upenn.edu/immun/koretzky.html>
To view Figure 2, please click on the following website: http://www.hhmi.org/lectures/biointeractive/animations/tcell/tcell_frames.htm
Fig. 2 The animation summerizes the importance of ITAM
and ZAP-70 in propagating the signal onward. In order to be activated,
the molecules contributing to the transduction cascade need to be present
together in the correct part of the cell. The inactivated molecule ZAP-70
(zeta-associated protein-70), a key player in the cascade, may randomly bump
into the ITAM subunit of the T-cell receptor. However, nothing will come of
these contacts unless ITAM is in the activated state. The binding of the APC to
the T-cell receptor triggers a series of events leading to ITAM activation.
(There was no email contact listed to request for permission to use the
animation.)
Mutations of ZAP-70 and Related Disorders
Patients who make a defective form of
the cytosolic protein tyrosine kinase ZAP-70, which transmit signals from the T
cell receptor, their CD4 T cells emerge from the thymus in normal numbers,
whereas CD8 T cells are absent. However,
the CD4 T cells that mature fail to respond to stimuli that normally activate
via the T cell receptor; hence, cause immunodeficiency (Janeway 2001).Thymocytes
expressing both CD4 and CD8 (double positive) are present while CD4- CD8+
thymocytes are absent, indicating that ZAP-70 is indispensable for the
development of CD8 single positive cells. Lack
of ZAP-70 may be partly compensated by the presence of syk, allowing the
development of CD4+ T cells but not CD8+ T cells (Noraz et al., 2000).
However, CD4+ T cells fail to respond to anti-CD3, mitogens or allogenic
cells in vitro, indicating a defective signal transduction and the activity of
natural killer (NK) cell is normal. Researches
show that mice with defective ZAP-70 lack both CD4+ and CD8+ T-cells in
peripheral lymphoid organs, which suggest that the requirement of ZAP-70 and syk
for T-cell development is different between mice and human ( Hivroz and Fischer,
1994). In addition, patients with
chronic lymphocytic leukemia (CLL) have low-level expression of CD38 and do not
express a detectable amount of ZAP-70 protein.
Leukemia cells from identical twins with CLL were found discordant for
expression of ZAP-70, suggesting that B-cell expression of ZAP-70 is not
genetically predetermined (Chen et al., 2002).
A mutation in ZAP-70 can result in
severe combined immunodeficiency (SCID), which is characterized by the absence
of both cellular and humoral immunity. SCID
mutation lacks appropriate rearrangements of TRC and Ig which results in absence
of mature T and B cells, and abnormal sensitivity to ionization radiation which
causes DNA double-strand to break. Intensive
researches demonstrate that ZAP-70 is essential for human T cell function and
suggest that CD4+ and CD8+ T cells depend on different intracellular signaling
pathways to support their development or survival (Elder et al., 1994). One
clinical case showed that T cells in ZAP-70 deficient patients are assumed to
have no helper functions for B-cell immunoglobulin synthesis.
The particular patient had immunoglobulin E (IgE) antibodies specific to
food allergens and the scientists investigated the mechanisms of switching to
IgE. It was found that the
peripheral blood mononuclear cells from the patient did not proliferate upon
stimulation with the antigens but produced distinct levels of IL-4.
Cell sorting analysis indicated that the cells that produced IL-4 in
response to the antigens were enriched in CD4+ T cells.
Purified CD4+ T cells from that patient expressed CD40L upon stimulation
with anti-CD3 and induced mature epsilon transcript on naďve B cells.
The results demonstrated that there was sufficient T cell receptor
signaling remained to exert antigen-specific IgE switching on B cells (Toyabe et
al., 2001).
Other related defects that cause SCID include:
1.
X-linked SCID-defects in common y chain for interleukin 2 (IL-2), IL-4,
IL-7,
IL-9
and IL-15 receptors.
2.
Adenosine deaminase (ADA) deficiency- defect in the enzyme of
interconversion pathways of purine metabolism which leads to the accumulation of
deoxyadenosine.
3.
Purine nucleotide phosphorylase (PNP) deficiency- different mutations in
PNP gene on chromosome 14 causes central nervous system of hypotonia, spasticity,
neutropenia or megaloblastic anemia.
4.
JAK3 kinase deficiency- mutation of JAK3 kinase in T-cell signal
transduction pathway is a rare cause of SCID.
5.
Reticular dysgenesis- the most severe type of SCID due to an inherited
defect in a pluripotential bone marrow stem cell.
6.
RAG defects- prevents VDJ rearrangement of immunoglobulin and T cell
receptor genes.
7.
MHC class II deficiency- caused by genetic defects in transcription
factors and fails to express MHC class II on lymphocytes and macrophages.
8.
Omenn’s syndrome- characterized by thickened eczematous skin, massive
lymphadenopathy and hepathosplenomegaly due to infiltration of polyclonal
lymphocytes, histiocytes, and eosinophils.
9.
Cellular immune defects and dwarfism- characterized by short limb
dwarfism, thin hair and variable T-cell deficiency with severe hypoplastic
anemia.
10.
Defects
in NF-AT transcription factor- defects of NF-AT do not have the capability to
control the transcription of many cytokines (Delves and Roitt, 1998).
Related Treatments
Even though the disease is extremely rare, nearly all patients with ZAP-70
defects presented with typical features of SCID in early life: severe pulmonary
infection often sustained by opportunistic pathogen (Pneumocystis carinii),
chronic diarrhea, failure to thrive, and persistent candidiasis.
ZAP-70 deficiency is ultimately fatal unless patients undergo bone marrow
transplantation (BMT) (Barata et al., 2001).
Patients with SCID can be treated with intravenous immunoglobulin
infusions at regular intervals and prophylaxis for pneumocystic carinii.
Bacterial or fungal infections are treated with intravenous infusions of
antibiotics or antifungal agents. Epstein-Barrvirus,
herpes virus and cytomegalovirus infection may result in systemic and lethal
disorders, hence it can be treated with antiviral reagents.
As for cytomegalovirus patients, a prophylactic infusion of
immunoglobulin is another treatment option.
Bone marrow transplantation (BMT) is the most favorable treatment to
reconstitute the impaired immune system in SCID (Taylor et al., 1996).
Chemotherapy, total body radiation, and antilymphocyte globulin can be
used to improve engraftment (Otsu et al., 2002).
HLA-haploidentical marrow from a parent is one of the primary sources for
BMT. Adenosine deaminase (ADA)
deficiency patients have two possibilities: enzyme replacement with polyethylene
glycol-modified ADA (PEG-ADA) or gene therapy inserted in a retroviral vector
(Delves and Roitt, 1998).
Alterations of ZAP-70 signaling
No drugs are currently known to affect the ZAP-70 protein.
However, drug targeting of single SH2 domain within ZAP-70 and Syk are
comparable in their abilities to mediate hematopoietic antigen receptor function
(Kong et al., 1995). An experiment
reported that Herpesvirus saimiri, which does not code for a ZAP-70 homologue,
can replace this tyrosine kinase because H. saimiri is an oncogenic virus that
has the ability to transform human T cells to stable growth based on mutual CD2
and CD3-mediated activation. In
the ZAP-70 deficient patient’s cell lines, CD2 and CD3 activation were
restored in terms of (Ca++), MAPK activation, cytokine production, and
proliferation. The transformed
cells expressed a high level of ZAP-70 related kinase Syk; therefore, it was
concluded that wild type H. saimiri can restore CD2 and CD3-mediated activation
in signaling deficient human T cells (Meinl et al., 2001).
References
Barata, L.T.,
R. Henriques, C. Hivroz, E. Jouanguy, A. Paiva, A.M. Freitas, H.B.Coimbra, A.
Fischer, and H.C. da Mota. 2001.
Primary
immunodeficiency
secondary to ZAP-70 deficiency. Acta
Med Port 14(4):413-7.
Blanchard, N.,
V. Di Bartolo, and C. Hivroz. 2002.
In the immune synapse, ZAP-70
controls T cell polarization and recruitment of signaling
proteins but
not formation of the synaptic pattern. Immunity
17(4):389-99.
Chen, L., G.
Widhopf, L. Huynh, L. Rassenti, K.R. Rai, A. Weiss, and T.J. Kipps.
2002. Expression of ZAP-70 is associated with increased B
cells receptor
signaling in chronic lymphocytic leukemia.
Blood 100(13):4609-14.
Cloning an Army
of T Cells for Immune Defense. < http://www.hhmi.org/lectures/biointeractive/animations/tcell/tcell_frames.htm>
Accessed 2003 17 Feb.
Elder, M.E., D.
Lin, J. Clever, A.C. Chan, T.J. Hope, A. Weiss, and T.G. Parslow. 1994.
Human severe combined immunodeficiency due to a
defect in
ZAP-70, a T cell tyrosine kinase. Science
264(5165):1596-9.
Eischen, C.M.,
B.L. Williams, W. Zhang, L.E. Samelson, D.H. Lynch, R.T. Abraham, and P.J.
Leibson. 1997. ZAP-70
tyrosine kinase is required for the up-regulation of Fas ligand in
activation-induced T cell apoptosis. Journal
of Immunology 159(3):1135-9.
Folmer, R.H.,
S. Geschwindner, and Y. Xue. 2002. Crystal
structure and NMR studies
of the apo SH2 domains of ZAP-70: two bikes rather than a tandem.
Biochemistry 41(48):14176-84.
Gelfand, E.W.,
K. Weinberg, B.D. Mazxer, T.A. Kadlecek, and A. Weiss.
1995. Absence of ZAP-70 prevents signaling through the antigen
receptor on
peripheral blood T cells but not on thymocytes.
Journal of Exp Med 182(4):1057-65.
Hatada, M.H.,
X. Lu, E.R. Laird, J. Green, J.P. Morgenstern, M. Lou, C.S. Marr, T.B. Phillips,
M.K. Ram, and K. Theriault. 1995. Molecular
basis for
interaction of the protein tyrosine kinase ZAP-70 with the T cell receptor.
Nature 376(6544):17-8.
Janeway, A.C.,
P. Travers, M. Walport, J.M. Shlomchik. 2001. Immunobiology: The
Immune System in Health and Disease. New York, NY: Elsevier Science
Ltd./ Garland Publishing, pp. 467, 1275, 1279, 2173, 2324.
Kong, G.H., J.Y.
Bu, T. Kurosaki, A.S. Shaw, and A.C. Chan.
1995. Reconstitution of Syk function by the ZAP-70 protein tyrosine
kinase.
Immunity
2:485-92.
Koretzky, G.
Molecular mechanisms of lymphocyte activation. <http://www.med.upenn.edu/immun/koretzky.html>
Accessed 2003 13 Mar.
Meinl, E., T.
Derfuss, R. Pirzer, N. Blank, D. Lengenfelder, A. Blancher, F. Le Deist, B.
Fleckenstein, and C. Hivroz. 2001.
Herpesvirus saimiri replaces ZAP-70 for CD3 and CD2-mediated T cell
activation. Journal of Biol Chem
276(40):36902-8.
Noraz, N., K.
Schwarz, M. Steinberg, V. Dardalhon,
C. Rebouissou, R. Hipskind, W. Friedrich, H. Yssel, K. Bacon, and N. Taylor.
2000.
Alternative
antigen receptor (TCR) signaling in T cells derived from ZAP-70-deficient
patients expressing high levels of Syk. Journal
of
Biol Chem
275(21):15832-8.
Otsu, M., M,
Steinberg, C. Ferrand, P. Merida, C. Rebouissou, P. Tigerghien, N. Taylor, F.
Candotti, and N. Noraz. 2002.
Reconstitution of
lymphoid
development and function in ZAP-70-deficient mice following gene transfer into
bone marrow cells. Blood
100(4):1248-56.
Pelosi, M. V.
Bartolo, V. Mounier, D. Mege, J.M. Pascussi, E. Dufour, A. Blondel, and O. Acuto.
1999. Tyrosine 319 in the interdomain B of ZAP-70 is a binding site
for the Src homology 2 domain of Lck. Journal
of Biol Chem 274:14229-37.
Pivniouk, V.,
and R.S. Geha. 2000. The role
of SLP-76 and LAT in lymphocyte development.
Current Opinion in Immunology 12:173-178.
Taylor, N., et
al. Immunomodulation et
immunotherapie. <http://www.igm.cnrs-mop.fr/pagedesequipes/webNTL.htm>
Accessed 2003 17 Feb.
Toyabe, S., A.
Watanabe, W. Harada, T. Karasawa, and M. Uchiyama.
2001. Specific
immunoglobulin E responses in ZAP-70-deficient
patients
are mediated by Syk-dependent T cell receptor signaling. Immunology
103(2):164-71.
Whisler, R., M.
Chen, B. Liu, and Y. Newhouse. 1999.
Age-related impairments in
TCR/CD3 activation of ZAP-70 are associated with reduced
tyrosine
phosphorylations of zeta chains and p56lck/p59fyn
in human T cells. Mechanisms
of Ageing and Development 111:1:49-66.
Davidson
Immunology Home Page
Davidson
College Home Page
Questions
or Comments, please email Michele Ho