This web page was produced as an assignment for an
undergraduate course at Davidson College.
Systematic Lupus Erythematosus
Introduction
SLE Autoantibodies
Etiology of SLE
A Possible Receptor-Editing
Defect
Polyclonal B Cell Activation
Antigen Driven B
Cell Activation
A Generalized
T Cell Tolerance Defect
Spontaneous
Activation of Autoreactive T Cells
A Possible Superantigen
Binding Site
Effects Of SLE Antibodies
Treatments
References
INTRODUCTION
Systematic lupus erythematosus (SLE) is an autoimmune
disease characterized by the presence of anti-DNA autoantibodies. The fact
that mammalian DNA is not immunogenic makes SLE an immunological mystery
(Zouali 1994). Victims of SLE may have skin rashes, but the real danger
of the disease is its direct attack of specific tissues and organs. Ultimately
the pathogenic autoantibodies bind to self proteins and form immune complexes
that cause severe organ damage (Blatt et al., 1999).
SLE AUTOANTIBODIES
Autoantibodies found in individuals with SLE are
primarily IgG1 and IgG3 immunoglobulin (Maddison 1999). The autoantibodies
are found in high concentrations and bind antigen with a high affinity.
A high frequency of positively charged and polar uncharged amino acids
are found in the CDRs of autoantibodies of SLE patients. Positively
charged amino acids like lysine, arginine, and histidine are able to interact
with phosphate residues of the DNA backbone. Polar, uncharged amino
acids like glutamine, glycine, and asparagine can bind to nucleic acids
(Zouali 1997). Since SLE autoantibodies favor DNA binding, they often attack
intracellular nucleoprotein particles such as nucleosomes and spiceosomes,
SLE autoantibodies also bind ribonucleoproteins such as Ro and La.
Ro is a 60 kD protein that forms complexes with small cytoplasmic RNA species.
La complexes with a variety of RNAs including the precursor forms of tRNA
(Maddison 1999). Crystallographic and binding analyses of single
stranded DNA:DNA complexes show that the single stranded DNA binding site
is a long cleft with a 1.2 nm gap between the heavy and light chain variable
regions. The single stranded DNA ligand is stabilized by thymine
bases of binding cleft aromatic side chains. High resolution structures
of anti-double stranded DNA:DNA complexes are currently unavailable (Blatt
et al., 1999).
ETIOLOGY OF SLE
Evidence suggests that SLE has a multi-factorial
etiology. Sex hormones, environmental factors, immunological dysfunction,
and genetics have all been hypothesized to be potential causes of SLE (Balow
et al., 2000). 90% of SLE affected individuals are women. And
the onset of SLE in women occurs most often during child-bearing years.
This data has led scientists to hypothesize that estrogen levels during
pregnancy may be a factor in the development of SLE. Researchers
have also noted that flares of SLE have been correlated with ultraviolet
light photosensitivity, which may mean that environmental agents have a
role in the onset of SLE (Blatt et al., 1999). However, the most
interesting theories about the etiology of SLE deal with defects of specific
elements of the immune system.
B cells originate from pluripotent hematopoetic stem
cells in the bone marrow and undergo several developmental stages before
expressing immunoglobulin specific for a single antigen. The specificity
of the immunoglobulin is determined by rearrangement of light chain and
heavy chain genes. In a healthy individual, immature B cells undergo
a "negative selection" process during which B cells that react to
self proteins are deleted, inactivated, or undergo further gene arrangement.
Immature B cells that recognize multivalent self antigens are eliminated
through clonal deletion. Immature B cells that bind to soluble self
molecules become anergic. In some cases receptor editing replaces
a self-reactive immunoglobulin rearrangement with a successive rearrangement.
Theoretically, if the immune system is working properly it should not produce
B cells with immunoglobulin that react with self proteins such as DNA.
However, individuals with SLE produce antibodies that react with self proteins
(Janeway et al., 1999). Researches have hypothesized that individuals
with SLE may have a defect in the negative selection process during B cell
development (Bensimon et al., 1994).
A POSSIBLE
RECEPTOR-EDITING DEFECT
One model of SLE focuses on a flaw in the receptor-editing
mechanism of B cells. Scientists examined anti-DNA antibodies of individuals
with SLE and noticed that SLE autoantibodies express only downstream Vk
genes. The kappa light chain variable region of an antibody molecule is
constructed from a variable (V) gene segment and a joining (J) gene segment.
There are about 30 Vk gene segments and 5 Vk genes that can be combined
during immunoglobulin rearrangement (Janeway et al., 1999). However, there
is currently no mechanism explaining which genes will be selected during
rearrangement. Usually, downstream Vk genes are selected during primary
rearrangement. If the immunoglobulin reacts with self protein it
can be saved from deletion by secondary rearrangements called receptor-editing.
Research has shown that during successive rearrangements upstream Vk genes
are utilized. The fact that SLE autoantibodies express only downstream
Vk genes indicates that these autoantibodies do not undergo secondary rearrangements.
Furthermore, secondary light chain rearrangements seem to favor downstream
Jk exons. However, SLE autoantibodies show no preference in expressing
downstream Jk exons. This information suggests that anti-DNA antibodies
do not undergo receptor-editing during the negative selection process.
Studies done on mice indicate that receptor-editing is critical in deleting
potentially pathogenic SLE anti-DNA antibodies. Therefore, individuals
with SLE may have defects in the receptor-editing process, resulting in
the failure to delete self-reactive antibodies (Bensimon et al., 1994).
POLYCLONAL
B CELL ACTIVATION
The fact that self-reactive B cells were not clonally
deleted during their development leads to complications down the road.
SLE patients have naïve B cells with anti-DNA antibodies circulating
throughout the body. There are two theories concerning how these
B cells are activated. The first theory suggests that polyclonally
activation of these B cells is responsible for the autoimmune response
of SLE. DNA present at high concentrations stimulate the SLE B cells
to proliferate and secrete antibody. However, immunologists do not
understand why polyclonal activation in SLE patients targets a specific
subset of autoantibodies (Zouali 1997).
ANTIGEN
DRIVEN B CELL ACTIVATION
While it is possible that SLE B cells are polyclonally
activated, SLE B cells have a pattern of mutations that suggest that an
antigen-driven response also plays a role in their activation. Analysis
of pathogenic SLE autoantibodies have revealed that these self-reactive
antibodies have a high rate of mutations clustered in hypervariable regions
(Zouali 1997). Hypervariable regions are segments of the heavy and
light chain V regions that contain a high degree of sequence variability.
The antigen-binding site of antibodies is composed of these hypervariable
regions, or complementarity-determining regions (CDRs). The high
variation of amino acids found in CDRs accounts for the antigen specificity
of the immune system. When a B cell binds antigen it moves to the
thymus where it is stimulated to differentiate and proliferate by the appropriate
helper T cell. Proliferating B cells move to a primary focus and
form a germinal center. In the germinal center the B cells undergo
somatic mutation. Ultimately, only somatically mutated B cells that
have the ability to bind antigen better than the original antibody bound
to the follicular dendritic cells survive. Therefore, somatically
mutated B cells accumulate mutations in CDRs (Janeway et al., 1999).
Some current theories hypothesize that polyclonally activated B cells are
responsible for producing SLE autoantibodies. However, SLE autoantibodies
have mutations clustered specifically in hypervariable regions, and SLE
antibodies have a high affinity for antigen. High affinity for antigen
and clustered mutations are characteristic of an antigen-driven response,
whereas limited random variations are typical of polyclonal activation
(Zouali 1997).
A GENERALIZED
T CELL TOLERANCE DEFECT
The human immune system is designed to delete or
inactivate self-reactive B cells before they leave the bone marrow.
The immune system of an individual with SLE malfunctions, and B cells that
bind self proteins evade detection and escape into circulation. However,
B cells do not act alone. Naïve B cells that bind antigen must
be stimulated by the appropriate T cell before they can differentiate and
proliferate. A healthy human immune system also has self-reactive "checks"
for T cells. T cells that are specific for self antigens are deleted
in the thymus (Janeway et al., 1999). One theory for the existence of autoreactive
T cells is that individuals with SLE may have an additional flaw in the
negative selection mechanism for T cells. However, a study conducted with
(New Zealand black (NZB) x New Zealand white (NZW))F1 (NZB/W) mice indicates
that the loss of T cell tolerance characteristic in SLE does not result
from a generalized defect in T cell tolerance. Instead, T cell tolerance
is caused by a more subtle defect in the immune system, such as abnormal
activation of T cells that are specific for a for a subset of autoantigens
(Wither et al., 2000).
SPONTANEOUS
ACTIVATION OF AUTOREACTIVE T CELLS
Current studies suggest that autoantibody Ig may
spontaneously activate T cells. Evidence shows that the heavy chain variable
region of autoantibodies stimulate spontaneous T cell activation in young
lupus-prone mice, whereas nonautoantibodies with the same heavy chain variable
region failed to stimulate T cell activation. The activated T cells can
then stimulate naive B cells expressing the self-reactive proteins.
The stimulated B cells proliferate and begin secreting SLE autoantibodies.
Evidence shows that these self-reactive T cells also recognize several
different self-Ig peptides. Autoimmune T cell recognition may diminish
over time resulting in a T cell repertoire that binds many self-Ig peptides
(Singh et al., 1998).
A POSSIBLE
SUPERANTIGEN BINDING SITE
SLE B cells produce a high number of autoantibodies
even though DNA is a weak immunogen. This observation has led researchers
to hypothesize that SLE autoantibodies may contain novel immunoglobulin
structural domains such as superantigen binding sites. Murine research
has revealed that SLE autoantibodies contain specific conserved nucleic
acid sequences at the D-J junction of the heavy chain variable region.
The location of these sequences suggests that this region may encode a
three-dimensional solvent-exposed determinant. The three-dimensional
solvent-exposed determinant, which is distinct from the classical antigen
binding site, may function as a superantigen or autoantigen binding site.
Further research is needed to determine what antigens bind to this alternative
binding site and the mechanism of B cell activation (Zack et al., 1994).
EFFECTS OF SLE
ANTIBODIES
DNA is almost exclusively intracellular. Only
a small amount of DNA from rupturing cells is found circulating in the
blood. However in individuals with SLE high quantities of DNA are present
in the blood serum. SLE autoantibodies bind circulating DNA and form antibody:antigen
immune complexes. Large amounts of immune complexes are continuously
produced. Many times these complexes become trapped in tissues of
blood vessels, joints, kidneys and brain. Immune complex deposits
activate phagocytic cells that cause tissue damage. More DNA and
nucleoproteins are released as tissue damage progresses, and more immune
complexes are formed (Janeway et al., 1999). Complement activity
amplifies the inflammatory reaction (Balow et al., 2000). Immune
complexes commonly deposit in the renal glomerulus and glomerular basement
membrane (Fig 1) causing proliferative and membranous forms of lupus nephritis
(Fig 2) (Janeway et al., 1999).
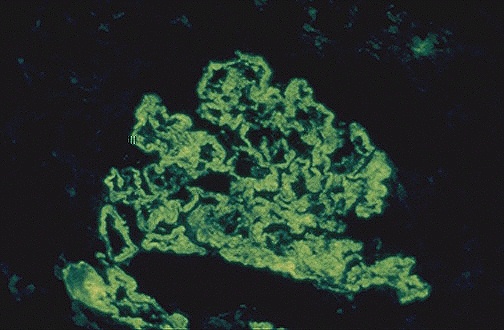
Figure 1. The granular pattern of immunofluorescence indicates the
deposition of immune complexes in the
basement membranes of the glomerulus. (Internet Pathology Laboratory
for Medical Education 2000)
Link
to The Internet Pathology Laboratory for Medical Education
Permission Pending.
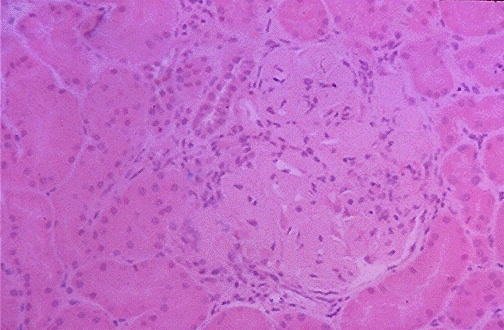
Figure 2. This image shows thickened capillary loops of a glomerulus
of an individual with lupus nephritis.
(Internet Pathology Laboratory for Medical Education 2000)
Link
to The Internet Pathology Laboratory for Medical Education
Permission Pending.
TREATMENTS
There are currently several treatments available
for SLE. Research has shown that testosterone suppresses B cell hyperactivity
in individuals with SLE. However, androgen receptors have yet to be found
on peripheral blood B cells indicating that testosterone may not act directly
on B cells. Studies show that testosterone therapy results in suppression
of anti-DNA antibodies (Kanda et al., 1997).
Since complement activity can lead to dangerous
consequences including inflammation, one experimental treatment has focused
on blocking terminal complement activity. The monoclonal antibody
to C5b blocks formation of membrane attack complexes. Murine studies
utilizing anti-C5b have resulted in less severe cases of lupus nephritis
(Balow et al., 2000).
Cytotoxic immunosuppressives such as cyclophosphamide
and azathioprine have proved to be effective in the treatment of lupus
nephritis. Both cyclophosphamide and azathioprine act by inhibiting
the proliferation of rapidly dividing B cells. However, both these
drugs are also toxic to cells of the bone marrow and gastrointestinal tract,
which result in unwanted side effects (Blatt et al., 1999).
REFERENCES
Balow JE. Boumpas DT. Austin HA. 2000. New prospects for treatment of
lupus nephritis. Seminars in Nephrology 20: 32-39.
Bensimon C, Chastagner P, Zouali M. 1994. Human lupus anti-DNA
autoantibodies undergo essentially primary Vk gene arrangements. European
Molecular Biology Organization Journal 13: 2951-2962.
Blatt NB, Glick GD. 1999. Anti-DNA autoantibodies and systematic lupus
erythematosus. Pharmacology and Therapeutics 83: 125-139.
The Internet Pathology Laboratory for Medical Education. 2000
Apr 14. The Internet Pathology Laboratory for Medical Eduation Homepage.
<http://www-medlib.med.utah.edu/WebPath/webpath.html#MENU>. Accessed
2000 Apr 21.
Janeway CA, Travers P, Walport M, Capra JD. 1999. Immunobiology:
the immune system in health and disease. New York, NY: Elsevier Science
Ltd/Garland Publishing. p. 209-216, 398, 499-500.
Kanda N, Tsuchida T, Tamaki K. 1997. Testosterone suppresses anti-DNA
antibody production in peripheral blood mononuclear cells from patients
with systematic lupus erythematosus. Arthritis and Rheumatism 40: 1703-1711.
Maddison PJ. 1999. Autoantibodies in SLE: Disease Associations. Advances
in Experimental Medicine and Biology 445: 141-145.
Singh RR, Hahn BH, Tsao BP, Ebling FM. 1998. Evidence for multiple mechanisms
of polyclonal T cell activation in murine lupus. The Journal of Clinical
Investigation 102: 1841-1849.
Wither J, Vukusic B. 2000. T cell tolerance induction is normal in the
(NZBxNZW)F1 murine model of systematic lupus erythematosus. Immunology
99: 345-351.
Zack DJ. Wong AL, Weisbart RH. 1994. Novel structural features of autoantibodies
in murine lupus:
a possible superantigen binding site. Immunology and Cell Biology 72:
513-520.
Zouali M. 1994. Human autoantibodies and their genes. Applied Biochemistry
and Biotechnology 47: 135-141.
Zouali M. 1997. The structure of human lupus anti-DNA antibodies. Methods:
A companion to Methods in Enzymology 11: 27-35.
Return
to Home Page
Return
to Davidson College Immunology Page

© Copyright 2000 Department of Biology,
Davidson College, Davidson, NC 28036
Send comments, questions, and suggestions to:
kafritchie@davidson.edu