This web page was produced as
an assignment for an undergraduate course at Davidson College.
|
Autoimmune Hemolytic Anemia
|
Introduction
Autoimmune Hemolytic Anemia
(AIHA) is the oldest recognized autoimmune deficiency in humans (Meyer
et al. 1998). It is one of many types
of anemias While red blood cells (RBCs) usually
circulate for 120 days before the spleen removes them from circulation,
in AIHA erythrocytes are prematurely destroyed by RBC autoantibodies.
When the bone marrow is unable to compensate for this hemolysis of RBCs
through hemopoiesis (production of new RBCs) a person is said to have hemolytic
anemia. AIHA may be acute or chronic and is sometimes fatal.
Women are twice as likely to have AIHA than are men (Kennedy
2000). However, in children the disease appears more often in
males and primarily affects children under 5 years of age as an acute hemolysis
(Gibson 1998). While AIHA affects only .001% of the general human
population (Hashimoto 1998), it is the most commonly occurring autoimmune
disease in canines (Day 1999).
Pathogenesis
The mechanism for hemolysis depends upon the autoantibody idiotype.
Additionally, not all autoantibodies cause hemolysis. Affinity of
the autoantibody for a species specific antigen and the autoantibodies'
abilities to cause hemoagglutination influence severity of the disease
(Shibata et al 1991). AIHA is usually caused by IgG1, IgG3,
and IgM autoantibodies. Occasionally the autoantibodies may be IgA.
Hemolysis results from either activation of the classical complement pathway
by IgM, IgG1, IgG3, and IgA or from phagocytosis or antibody-dependent
cell cytotoxicity (ADCC) by Ig1 and Ig3 (Gibson 1998). Extravascular
phagocytosis and ADCC mediated hemolysis result from recognition of the
antibody by Fc receptors on macrophage, resulting in erythrophagocytosis,
or on K cells in the spleen, resulting in the release of lysosomal enzymes.
Studies conducted by Meyer et al suggest that IgG1 promotes erythrophagocytosis
via the FcyRIII receptor and that IgG2 may also participate in erythrophagocytosis,
but primarily through FcyRI (1998). In compliment mediated hemolysis, binding
of the antibody initiates the compliment cascade. The compliment
cascade may terminate at c3b formation, when macrophage, especially hepatic
Kupffer's cells, engulf the antibody coated erythrocyte. Continuation
of the complement cascade leads to formation of a membrane attack complex
and intravascular hemolysis. However the results of some studies
suggest that complement may not play as significant a role in hemolytic
anemia as is currently thought (Meyer et al 1998).
Partial extravascular hemolysis creates sperocytes,
which are spherical, rigid erythrocytes that have lost part of their cell
membrane. The cells are fragile and therefore easily damaged and
destroyed (Domen 1998). IgG hemolysis occurs largely in the spleen
while hemolysis by IgM occurs in the liver (Merck
2000). Additionally, because splenic macrophage possess both
complement and FcR receptors while hepatic macrophage display only complement,
most extravascular hemolysis occurs in the spleen (Gibson 1998).
Intravascular hemolysis leads to fragmentation of erythrocytes into helmet
shaped schizocytes. Peripheral blood smears can be examined for sperocytes
and schizocytes to provide information about the mechanism of a particular
patient's AIHA (see figures 2 and 3).
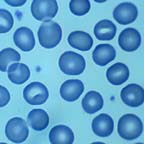
Normal Red Blood Cells
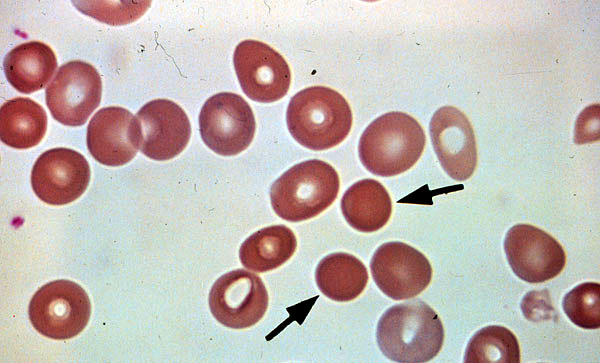
Sperocytes
Schizocytes
Peripheral blood smear.
Peripheral blood smear.
Schizocytes result from fragmentation of
Note the areas of central pallor.
Sperocytes formed from partial
erythrocytes in intravascular hemolysis.
extravascular hemolysis.
Note the lack of areas of central
pallor.

Figure
1
Figure 2
Figure 3
Image taken from Dr
Ed Uthman's
Image taken from KU
Pathology 851
Image taken from Dr.
Ed Uthman's
web page on hemolytic
anemia
image web page with permission of
web page on hemolytic animas with
with permission of author
(Uthman 2000) author (Woodroof 2000).
permission of author (Uthman 2000).
Types of AIHA
AIHA is a heterogeneous disease and includes
Warm AIHA (WAIHA), Cold AIHA (CAD), Paroxysmal Cold Hemoglobinuria (PCH),
and Drug Induced Hemolytic Anemias (DIHA); all of which are characterized
by production of autoantibodies to RBCs . DIHA are further subdived
into three classes, based upon the binding site of the autoantibody.
WAIHA and CAD may be either idiopathic or may exist secondary to another
autoimmune disease. Some patients have mixed AIHA, which manifests
both CAD and WAIHA autoantibodies. Table 1 summarizes some of the
characteristics of each AIHA discussed below.
WAIHA
WAIHA is the most commonly occurring form of
AIHA. Autoantibodies produced in WAIHA are non-specific and bind
to all RBCs at 37'C, with the exception of those which lack Rh antigen.
The autoantibodies are of polyclonal origin and usually bind to the Rh
antigen (Hashimoto 1998). Approximately 60% of WAIHA cases are idiopathic
(Smith 1999). Secondary WAIHA often occurs with chronic lymphocytic
leukemia and may also occur with diseases such as systematic lupus, solid
tumors, myloproliferative diseases, and hepatitis A. Hemolysis is
usually extravascular and occurs via partial phagocytosis or by ADCC.
The autoantibodies causing hemolysis are most frequently IgG1 and IgG3.
WAIHA autoantibodies are usually of polyclonal origin. However, in
cases where IgM and IgA predominate, the presence of autoantibody only
occasionally causes WAIHA (Gibson 1988).
CAD
In CAD, the autoantibody is usually monoclonal
IgM, but occasionally IgG or IgA, with kappa light chains.
The autoantibody binds best at temperatures below 4'C. However, the
thermal amplitude (temperature range within which the antibody binds) varies,
and higher thermal amplitudes tend to indicate a more severe form of the
disease (Hashimoto 1998). A polyclonal IgM has been reported as well,
but it is rarely pathological and reacts only at very low temperatures
(Domen 1998). The cold agglutinin antibody is specific for either
I antigen or i antigen. Idiopathic CAD usually occurs in adults,
especially the elderly, as a chronic, mild anemia. Because of the
antibody's temperature sensitivity, the condition worsens in winter and
binds when RBCs enter peripheral circulation (Smith 1999). This form
of CAD can cause both intravascular and extravascular hemolysis.
Secondary CAD frequently appears in children who have recently had viral
or bacterial infections such as Mycoplasma pneumoniae and infectious mononucleosis.
Unlike idiopathic CAD, this condition occurs suddenly and is acute.
However, the condition also tends to be transient (Hashimoto 1998).
CAD also exist in a chronic form when the disease occurs secondary to B-cell
lymphomas or chronic lymphocytic leukemia (Zilow et al 1994).
PCH
PCH is caused by an IgG biphasic autoantibody
which binds to RBCs at temperatures below 4'C. The autoantibody is
usually specific for the globoside glycosphingolipid P antigen (Rosenfield
and Diamond 1981). The first two components of the complement system
bind at 4'C , and the cascade is completed at 37'C. In the early
1900's PCH occurred mostly among syphilis victims as an acute disease.
Most cases today occur in children after infection with measles, mumps,
chickenpox or influenza (Smith 1999). This more recent condition
causes severe and rapid intravascular hemolysis that may be life threatening
for 10-14 days after onset (Rosenfield and Diamond 1981). However,
PCH is usually a self limiting form of AIHA.
Mixed AIHA
In mixed AIHA both warm agglutinate IgG and cold
agglutinate IgM autoantibodies are present. The autoantibodies may
or may not have specificity for I or i antigen. Both intravascular
and extravascular hemolysis are observed. Approximately 50% of mixed
AIHA are idiopathic, while secondary mixed AIHA commonly occurs in collagen
autoimmune diseases like lupus . This form of AIHA appears as a sudden,
acute disease but often becomes a chronic condition (Smith 1999).
Drug Induced AIHAs
While autoimmune hemolytic anemia is a rare disease,
the incidence of DIHA is increasing significantly. Over 70 different
drugs have induced either a positive Coombs' test or immune hemolysis (Wright
1999). Drugs have been observed to induce four types of autoantibody
binding to erythrocytes. However, only three of these types of binding
are known to cause hemolytic anemia. The characteristics of DIAHAs
resemble those of WAIHA. In the hapten mechanism, the drug binds
to an RBC which acts as a carrier for the drug hapten. In the immune-complex
mechanism the drug first binds to the antibody and the drug-antibody complex
then binds to the RBC. In the autoimmune mechanism, the autoantibody
binds directly to the RBCs. (Jefferies 1994). Drugs such as
cephalosporins have been shown to modify the RBC membrane.
Serum proteins such as immunoglobulins and complement proteins then bind
non-specifically to the RBCs. However, the weak binding of RBCs in
membrane modification has not yet been demonstrated to cause hemolysis
(Mueller-Eckhart and Salama 1990). Table 2 lists drugs and
the specific mechanism by which they induce hemolysis. Nonetheless,
not all drugs are specific to one particular mechanism. Certain drugs
have been found to elicit mixed responses; for instance, administration
of the drug, nonifensine, can induce both immune complex and autoantibody
mechanisms of hemolytic anemia (Petz 1993).
Table 1
Characteristics of Five Types of Autoimmune Hemolytic
Anemias (Compiled from Hashimoto 1998, Smith 1998)
c = complement, AuAb = autoantibody
| AIHA |
% of Cases |
Pathogenesis |
Predominating Blood Group |
Antibody Type |
DAT Results |
Antibody in Eluate |
Treatment Options |
| WAIHA |
80% |
AuAb bind RBC at 37 'C |
Rh |
IgG |
IgG, IgG+C, C(rare) |
IgG |
variable: cortecosteroids. immunosuppression, danazol,
IV gamma globulin |
| CAD |
20-25% |
AuAb bind to RBC at 4' C |
I, i |
IgM, IgG |
C3d |
IgM |
frequently not needed |
| AIHA Mixed |
7-8% |
broad amplitude of reactivity to 37 'C |
Possibly I, i |
IgM,IgG |
IgG, C3d |
IgG |
cortecosteroids |
| PCH |
1% |
AuAb binds RBC at 4 'C; fixes complement;complement cascade
completed at 37 'C |
P |
IgG |
C |
nonreactive |
self-limiting,; possibly transfusion |
| DIHA |
12-18% |
AuAb binds drug, or binds drug then RBC, or binds drug-rbc |
--- |
--- |
--- |
--- |
discontinue drug, occasionally transfusion |
Table 2
Partial list of drugs reported to cause AIHA
(Jefferies 1994)
| Hapten Mechanism |
Penicillin, Cephalothin, Ampicillin, Carbenicillin, Methicillin,
Cephaloridine |
| Immune Complex Mechanism |
Quinine, Quinidine, Rifampin, Antihistamines, Sulfonamides,
Tetracyclin, Insulin, Streptomycin, Acetaminophen, Cephaosporin, Dipyrone,
Isoniazid, Tolmetin |
| Autoantibody Mechanism |
a-Methyldopa, L-Dopa, Ibuprofen, Procainamide, Thioridazine |
Diagnosis
Many symptoms of AIHA
resemble those of other anemias and include nosebleeds, bleeding gums,
chills, fatigue, paleness, shortness of breath, and jaundice (Kennedy
2000). Symptoms of AIHA may also include an enlarged spleen,
due to excessive RBC destruction, and dark urine, due to an excess of unprocessed
catabolites resulting from RBC hemolysis. Patients with CAD may experience
numbness and pain in cooler temperatures as a result of cyanosis
(Domen 1998). Because the bone marrow attempts to compensate for
the loss of RBCs through elevated hemapoiesis laboratory test results such
as high reticulocyte (developing RBCs) counts are suggestive of a
hemolytic anemia. Click here
to view a map of diagnostic procedures to identify various types of anemias.
After hemolytic anemia has been diagnosed clinical
history, Coombs' test (direct antiglobulin test or DAT), and blood smear
morphology aid in determination of its origin (Uthman
2000). DAT is the most important assay for distinguishing AIHA
from other types of hemolytic anemias (Jefferies 1994). A DAT to
determine the presence of either c3 or IgG bound to erythrocytes is performed
and a positive test results in erythrocyte agglutination (Figure 4).
After the initial positive DAT, additional DATs are conducted to determine
whether c3, IgG or both proteins are binding to the RBC. In CAD,
only c3 binding will usually be detected when the test is conducted at
temperatures around 37'C, but in WAIHA the DAT may be positive for IgG
alone or both IgG and c3. If IgG is detected, the autoantibody
may be eluted and tested for antigen
specificity,
especially when cross-matching for a transfusion.
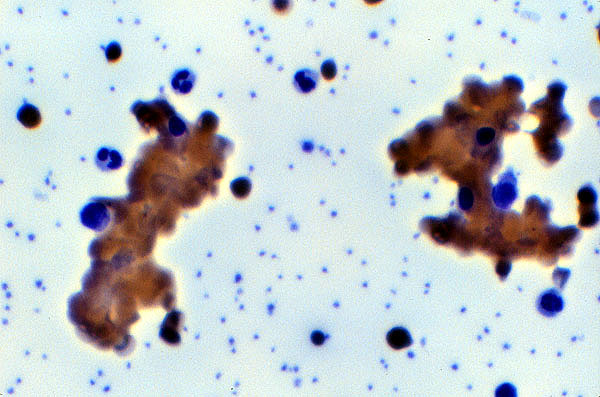
Figure 4
Peripheral smear
RBC agglutination caused by cold
agglutination autoantibody.
Image taken from KU
Pathology 851 image web page with permission of author (Woodroof 2000).
AIHA is a drug induced condition and tests against
drug-treated RBC can confirm the mechanism of the drug induced reaction
(Wright and Smith 1999). The Donath-Landsteiner test is preformed
to detect PCH. In this test, the IgG autoantibody is incubated with normal
RBC and serum at 4'C and then warmed to 37'C to cause hemolysis (Jefferies
1994).
Etiology
New Zealand Black mice (NZB) provide the current
animal model to study AIHA, while methyl-dopa drug induced AIHA has provided
researchers with a human model of both AIHA as well as autoimmune diseases
as a whole. Nevertheless, the etiology of AIHA is still not
understood.
Much research supports an antigen induction model
of AIHA. In mice, band 3, an erythrocyte anion exchange protein,
appears to be the predominate antigen for RBC autoantibodies. However,
not all autoantibodies binding band 3 produce pathological effects.
The protein appears to serve a natural role in the elimination of aged
RBCs; in aged RBCs, band 3 aggregates and antibody binds at higher density
to facilitate clearance of the cells (Diilulio et al 1997). However,
NZB mice with band 3 reactive CD4 T cells do produce pathogenic autoantibodies
(Perry et al 1996). In a study by Barker et al on humans, many AIHA
patients were also found to express T helper cells which bind to the Rh
antigen on human RBCs. B cells, however, were found not to react
with same epitopes on the Rh antigen which is recognized by the T helper
cells (1997). As a result of these studies, researchers have proposed
that induced changes in MHCII autoantigen processing results in the presentation
of previously cryptic epitopes to which naive Rh reactive T cells respond
(Barker et al 1997, Shen et al 1996). This theory is supported by
a study by Diiulio et al on NZB mice which found yet another autoantibody
for murine RBCs which binds to a partially masked epitope when the RBCs
are treated with protease to enhance expression of the epitope (1997).
The TH-1 predominated response to band 3 elicits IFN-Y production, and
this cytokine may promote presentation of the cryptic epitopes (Shen et
al 1996). However, how a self-reactive T cells might escape clonal
deletion to respond to the self-antigen is not yet understood.
Evidence from other studies supports a polyclonal
activation model instead of an antigen-induced model of AIHA. For
instance, Hernandez et al found RBC autoantibodies in both healthy and
AIHA individuals but that autoantibody levels were much higher in AIHA
individuals. Higher levels of RBC autoantibodies could be induced
by polyclonal activation (1990). Yet other research suggests B-1
involvement in AIHA. Unlike conventional self-reactive B cells in the periphery
and the bone marrow, self-reactive B-1 cells in the peritoneal cavity are
separated from RBCs and may therefore escape clonal deletion. Oral
administration of lipopolysaccharides (LPS) to HL mice with H and L chains
derived from NZB mice induces peritoneal B-1 cell secretion of RBC autoantibodies
in the gut lumen and results in AIHA (Nisitani et al 1997). In contrast
to other studies, this study also found that TH2 cells could cause AIHA
through Il-5 and Il-10 induction of autoantibody secreting B-1 cells.
Additionally, elimination of B-1 cells reduces not only the amount of IgM
autoantibody but also the amount of IgG autoantibody, demonstrating B-1
cell involvement in IgG production as well as IgM production (Murakami
et al 1995). As a result of the evidence for both polyclonal and
specific antigen-induced responses, a unifying model whereby antigen-induced
specific responses are preceded by polyclonal activation has been proposed
for all autoimmune diseases (Dziarski 1988).
Mueller and Eckhart have proposed yet another mechanism to explain DIHAs.
They suggest that, rather than through inhibition of T cell suppresser
function or a failure of immune tolerance, all forms of drug induced antibodies
are caused by the formation of a composite antigenic structure upon binding
of the drug or drug metabolites to a site on the RBC. Antibodies
elicited by an altered membrane structure can react with the drug, the
drug-RBC complex, and/or the RBC alone (1990).
Table 2
Possible Factors Contributing to the Etiology of AIHA
(Gibson 1998)
Potential Defects in Self Tolerance
-
clonal deletion/clonal anergy/clonal abortion
-
suppressor t cells
|
Development of Autoimmunity
-
altered-self antigen
-
abnormalities in antigen presentation
-
abnormalities in immunoregulation
-aberrations in helper and /or suppressor T cell number and function
-B cell hyper-reactivity
-
non-immunological/environmental factors
|
Disease-Specific Factors
-
eg infections, hormonal influences, drugs
|
There is no cure for AIHA.
Since many patients exhibit AIHA as a secondary disorder, patients are
examined for other underlying immune disorders. If found, treatment
of the primary disease often resolves AIHA as well. Conventional
treatments include corticosteroids, splenectomy, and cytotoxic agents.
The corticosteroid, prednisone, is frequently the first method of treatment
for WAIHA forms of AIHA, with about 80% to 90% response rate over a period
of weeks to months (Domen 1998). Prednisone acts as an anti-inflammatory
agent and binds to regulatory gene sequences and modulating transcription.
In AIHA, prednisone reduces the number of Fc receptors on macrophage, increases
autoantibody coated RBC survival, and moderately decreases the amount of
autoantibody produced (Gibson 1998). Splenectomy or cytotoxic agents
are often second line therapies for WAIHA. Removal of the spleen
reduces destruction of IgG coated RBCs and allows damaged cells to circulate
longer. Splenectomy is usually not an effective treatment in CAD
since most hemolysis of IgM coated cells occurs in the liver rather than
the spleen (Hashimoto 1998). Azathioprine and cyclophosphamide are
the most frequently used cytotoxic drugs in cases of AIHA. These
agents reduce autoantibody production and suppresses effector mechanisms
by preventing cell division and cytokine production (Smith 1999).
Azathioprine and cyclophoshamide are preferred for elderly patients, for
whom splenectomy presents greater risk (Gibson 1998). AIHA patients
also use folic acid supplements to aid in RBC maturation and replacement
of destroyed RBC.
Whether or not transfusions should be used to treat AIHA is still controversial.
Serological evaluations routinely done before blood transfusions are complicated
in AIHA, especially in the WAIHA form of the disease because special procedures
most be preformed to separate autoantibodies from alloantibodies before
an indirect agglutination test is performed (Jefferies 1994). Additionally
WAIHA antibodies may destroy transfused cells as rapidly as they destroy
self-RBCs, unless the transfused blood is Rh- (Smith 1999). CAD and
PCH present less risk but transfused cells may also be incompatible in
these forms of AIHA as well (Domen 1998). Additionally repeated transfusions
may increase the risk of alloimmune response. Thus some researchers
argue that the temporary benefits of transfusion are not warranted (Smith
1999, Gibson 1998). Other researchers argue that the transfusions
do not result in intensified hemolysis nor alloimmunuzation (Salama and
Berghofer 1992). However, when patients experience acute AIHA and
are at high risk for central nervous system or cardiac failure, transfusion
is warranted, even where blood has not been thoroughly cross-matched (Hashimoto
1998).
In addition to conventional treatment, more recent therapies have been
explored. Danazol, a modified androgen which reduces both the amount
of c3 bound to RBCs and the number of Fc receptors on macrophage, may alleviate
WAIHA (Eckman
1998). Cyclosporin A has also recently been used to successfully
treat WAIHA through inhibition of T cell activation and proliferation (Smith
1999). Trials with intravenous IgG immunoglobulins (IV-IgG) have
shown variable success (Domen 1998). IV-IgG anti-idiotypic immunoglobulins
appear to neutralize autoantibodies by forming idiotype-anti-idiotype complexes
to prevent coating of the RBC, bind B cell receptors to decrease autoantibody
production, and regulate T cell function (Choudry, Mahapatra, and Kashyap
1998). Another experimental treatment has effectively reduced hemolysis
through administration of monoclonal antibodies for the IgG Fc receptor
(Gibson 1998).
CAD does not respond well to many of the conventional treatments; however,
the disease can often be treated through supportive methods alone such
as by keeping the patient warm and by drinking lots of fluids. If
CAD or PCH is severe, transfusions or cytotoxic agents may be administered
(Gibson 1998). Plasmaphoresis to remove autoantibody is sometimes
effective in temporary treatment of CAD, but usually not WAIHA, because
at 37'C IgM is no longer bound to RBCs and is intravascularly distributed
(Gibson 1998). Because hemolysis occurs at decreased temperatures,
cardiac patients should be tested for CAD prior to surgery, as cold heart
surgery could lead to severe hemolysis (Hashimoto 1998). Conventional treatment
is usually not used in DIHA either; discontinuation of the reactive drug
usually resolves RBC hemolysis.
References
Barker, R., Hall, A., Standen, G., Jones, J., Elson, C. 1997.
Identification of T-Cell Epitopes on the Rhesus Polypeptides in Autoimmune
Hemolytic Anemia. 90 (7) : 2701-2715.
Choudhry, V., Mahaptra, M., Kashyap, R. 1998. Immunoglobulin
Therapy in Immunohematological Disorders. Indian Journal of Pediatrics.
65 (5) : 681-690.
Cornell University Medical School. 1996 Oct 3. Cornell Pathology
Image Collection-Classification of Anemias.
<http://edcenter.med.cornell.edu/CUMC_PathNotes/Hematopathology/3040.gif>
Accessed
2000 April 19.
Day, M. 1999. Antigen Specificity in Canine Autoimmune Haemolytic
Anaemia. Veterinary Immunology and Immunopathology. 69 (2-4)
: 215-224.
Diiulio, N., Fairchild, R., Caulfield, M. 1997. The Anti-Erythrocyte
Autoimmune Response of NZB Mice. Identification of Two Distinct Autoantibodies.
Immunology. 91 (2) : 246-251.
Domen, R. 1998. An Overview of Immune Hemolytic Anemias.
Cleveland Journal of Medicine. 65 (2) : 89-99.
Dziarski, R. 1988. Autoimmunity : Polyclonal Activation
or Antigen Induction? Immunology Today. 9 (11) : 340-342.
Eckman, J. 1998 April 14. Disorders of Red Cells.
<http://www.emory.edu/INT_MED_REV/Atlanta/paper/paper.htm>
Accessed 2000 April 15.
Gibson, J. 1998. Autoimmune Hemolytic Anemias : Current
Concepts. Australian and New Zealand Journal of Medicine. 18
(4) : 625-637.
Hashimoto, C. 1998. Autoimmune Hemolytic Anemia. Clinical
Reviews in Allergy and Immunology. 16 (3) : 285-295.
Hernandez-Jodra, M., Hudnall, S., Petz., L. 1990. Studies
of In Vitro Red Cell Autoantibody Production in Normal Donors and in Patients
with Autoimmune Hemolytic Anemia. Transfusion. 30 (5) :
411-416.
Jefferies, L. 1994. Transfusion Therapy in Autoimmune Hemolytic
Anemia. Transfusion Medicine. 8 (6) : 1087-1104.
Kennedy, R. 2000. The Doctors' Medical Library-Hemolytic
Anemia. <http://www.medical-library.net/sites/hemolytic_anemia.html
> Accessed 2000 April 7.
Merck. 2000 March 19. The Merck Manual of Diagnosis and
Therapy- Anemias Caused by Excessive Hemolysis. < http:/www.merck.com/pubs/mmanual/section11/chapter127/127d.htm
> Accessed 2000 April 10.
Meyer, D., Schiller, C., Westermann, J., Izui, S., Hazenbos, W., Verbeek,
J., Schmidt, R., Gessner, J. 1998. FcYRIII (CD16)-Deficient
Mice Show IgG Isotype-Dependent Protection to Experimental Autoimmune Hemolytic
Anemia. Blood. 92 (11) : 3997-4002.
Mueller-Eckhart, C., Salama, A. 1990. Drug-Induced Immune
Cytopenias : A Unifying Pathogenetic Concept with Special Emphasis
on the Rule of Drug Metabolites. Transfusion Medicine Reviews.
4 (1) : 69-77.
Murakami, M., Honjo, T. 1995. B-1 Cells and Autoimmunity.
Annals of the New York Academy of Sciences : Vol 764. Eds. Boland,
B, Cullinan, J., Kimball, C. New York : New York Academy of
Sciences. 402-409.
Nisitani, S., Murakami, M., Honjo, T. 1997. Anti-Red Blood
Cell Immunoglobulin transgenic Mice. An Experimental Model of Autoimmune
Hemolytic Anemia. Annals of the New York Academy of Sciences :
Vol 815. Eds. Boland, B, Cullinan, J., Kimball, C.
New York : New York Academy of Sciences. 246-252.
Perry, F., Barker, R., Mazza, G., Day, M., Wells, A., Shen, C., Schofield,
A., Elson, C. 1996. Autoreactive T Cell Specificity in Autoimmune
Hemolytic Anemia of the NZB Mouse. European Journal of Immunology.
26 (1) : 136-141.
Petz, L. 1993. Drug-Induced Autoimmune Hemolytic Anemia.
Transfusion Medicine Reviews. 7 (4) : 242-254.
Rosenfield, R., Diamond, S. 1981. Diagnosis and Treatment
of the Immune Hemolytic Anemias. Haematologia. 14 (3) :
247-256.
Salama, A., Berghofer, H. 1992. Red Blood Cell Transfusion
in Warm-Type Autoimmune Haemolytic Anaemia. Lancet. 340 (8834-8835)
: 1515-1526.
Shen, C., Mazza, G., Perry, F., Beech, J., Thompson, S., Corato, A.,
Newton, S., Barker, R., Elson, C. 1996. T-Helper 1 Dominated
Responses to Erythrocyte Band 3 in NZB Mice. Immunology. 89
(2) : 195-199.
Shibata, T., Berney, T., Reininger, L., Chicheportiche, Y., Ozaki, S.,
Shirai, T., Izui, S. 1991. Monoclonal Anti-Erythrocyte Autoantibodies
Derived from NZB Mice Cause Autoimmune Hemolytic Anemia by Two Distinct
Pathogenic Mechanisms. International Immunology. 2 (12) :
1133-1142.
Smith, L. 1999. Autoimmune Hemolytic Anemia : Characterization
and Classification. Clinical Laboratory Science. 12 (2) :
110-114.
Sullivan, J. 2000 Jan 29 Cells Alive- The Gallery.
< http://www.cellsalive.com/
> Accessed 2000 April 14.
Territo, M. 1997 Dec 8. Pathophysiology of Disease- Hematopathology-Anemia.
<http://www.pathnet.medsch.ucla.edu/med-edu/ppd/book/image30.gif>
Accessed 2000 April 14.
Uthman, E. 2000 March 18. Blood Cells and the CBC.
< http://www.neosoft.com/~uthman/blood_cells.html>
Accessed 2000 April 10.
Uthman, E. 2000 March 18. Hemolytic Anemias.
< http://www.neosoft.com/~uthman/hemolytic_anemia/hemolytic_anemia.html
> Accessed 2000 April 10.
Woodroof, J. 2000 Jan 10. Pathology 851 Blood and Lymphoid
Tissues I Supplemental Image Database. < http:/www.kumc.edu/instruction/medicine/pathology/ed/ch_20a/ch20a_nf.html>
Accessed 2000 April 10.
Wright, M., Smith, L. 1999. Laboratory Investigation of
Autoimmune Hemolytic Anemias. Clinical Laboratory Science.
12 (2) : 119-122.
Wright, M. 1999. Drug-Induced Hemolytic Anemias- Increasing
Complications to Therapeutic Interventions. Clinical Laboratory Science.
12 (2) : 115-118.
Zilow, G., Kirschfink, M., Roelcke, D. 1994. Red Cell Destruction
in Cold Agglutinin Disease. Infusionstherapie und Transfusionmedizin.
21 (6) : 410-415.
Return
to my Main Page
Davidson College
Immunology
Home Page.

Send comments, questions, and suggestions to: rawolf@davidson.edu
{kind=link}
{kind=link}