Dystrophin
This web page was produced as an assignment for an undergraduate course at Davidson College.
The Protein: Structure, Function, and Evolution
Dystrophin is a large cytoskeletal protein expressed primarily in skeletal muscle, cardiac muscle, and the brain (inneuronal cells). The dystrophin gene is a highly complex and large gene that enables a variety of dystrophin isoforms. Dystrophin is the largest known gene in nature (2.5 Mb), totaling roughly .1 % of the human genome (López et al., 1997). The gene comprises of 79 exons and at least eight different tissue-specific promoters that each code for different first exons. These promoters allow tissue specificity for different isoforms of dystrophin in the body. The gene also has two separate poly-adenylation sites to provide even more specification (Sugita, et al., 2001). Dystrophin expression begins during myogenesis. Its mRNA is 14 Kb long and translates into a protein 3,685 amino acids long, having a mass of 427 KDa (muscle). Smaller isoforms include a 260 KDa retinal protein, and a 140 KDa brain protein (López et al., 1997).
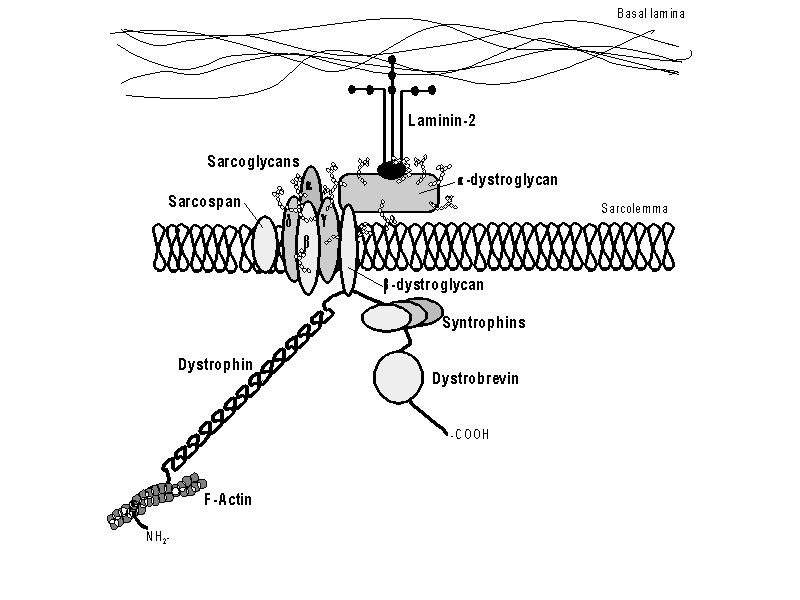
| Figure 1. The dystrophin-glycoprotein complex. The N-terminus of dystrophin attaches to F-actin and the C-terminus attaches to ß-dystroglycan. This entire complex provides support during muscle contractions. Permission pending from author, see source. |
The dystrophin protein is a central member of
the dystrophin-glycoprotein complex. This complex spans the cell membrane of
muscle cells to anchor the cytoskeleton to the extracellular matrix (Malcolm,
2001). In anchoring the cytoskeleton inside the cell, this complex is able
to provide shock absorption to reduce the mechanical stress muscle cells endure
with each contraction. Dystrophin circles the interior of muscles cells to give
added strength and flexibility. Dystrophin also helps muscle fibers to differentiate
into specific groups (Pestronk,
2002). ß-dystroglycan is a transmembrane protein that attaches to
the carboxyl terminus of dystrophin, connecting the intracellular dystrophin
to the extracellular a-dystroglycan. Dystrophin itself has no transmembrane
domains, only four cytoskeletal domains (Malcolm,
2001):
Domain 1: Actin Binding Domain
Domain 2: Rod Domain
Domain 3: Cysteine-Rich Domain
Domain 4: ß-dystroglycan Binding Domain
| Figure 2. Chime image of N-terminus actin binding domain of human dystrophin. This binding site links the cyctoskelton actin protein to dystrophin, which anchors actin to the dystrophin-glycoprotein complex and so to the extracellular matrix. Click Here for Source: PDB |
Dystrophin has homologies in its carboxyl terminus to utrophin and dystrobrevin. Both utrophin and dystrobevin attach to the dystrophin-glycoprotein complex or to similar complexes within the cell. One incredible aspect of dystrophin is its conservation throughout evolution of both structure and function. In sea urchins, the dystrophin gene is homologous to the smaller human isoform of dystrophin, Dp116 (Nudel, 2001). Drosophilia has three isoforms of dystrophin-like proteins: dmDLP1, dmDLP2, and dmDLP3. Drosophilia also has a small isoform of dystrophin, dm Dp186, which is homologous to the brain isoform of human dystrophin. The presence of larger dystrophin isoforms in muscle cells and a smaller isoform in the brain and nervous system remains highly conserved, even between such distant species as humans and flies (see figure 3).
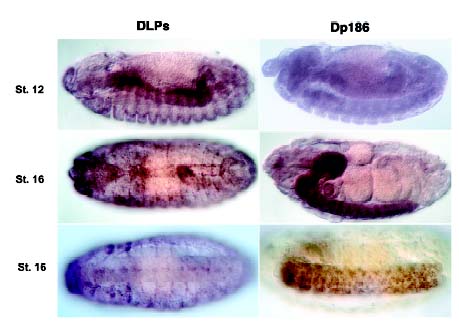
| Figure 3. In situ hybridization staining of the DLP's and Dp186 in drosophilia. The DLP's are the larger dystrophin-like proteins that are seen here associated with endoderm, segment borders, and pericardial cells in different stages of development. Dp186, much like the human brain isoform, is the smaller dystrophin isoform that is associated with the brain and central nervous system. Permission pending from author, see source. |
Muscular Dystrophy
Duchenne muscular dystrophy and Becker muscular dystrophy are the two main muscular dystrophies. Both diseases reduce motor function, cause mental retardation (lack of brain isoform), and reduced vision (lack of retinal isoform). When dystrophin is in small quantities or missing altogether in the cell, muscle fibers will go through cycles of muscle breakdown and regeneration until the muscle is replaced by connective tissue and fat (Malcolm, 2001). When dystrophin is not present, the muscle cells lack the shock absorption and support needed during contraction and eventually tiny holes tear in the cell membrane. The presence of these holes activates proteases that lyse the cellular contents, destroying the cells. Once the cells are dead they can be replaced by new muscle cells, however after continual periods of regeneration the tissues give up and the cells are replaced with connective tissue and fat.
Duchenne muscular dystrophy (DMD) is caused by a frameshift
mutation in the dystrophin gene 96 % of the time, leading to premature stop
codons (Pestronk,
2002). DMD is a recessive, fatal, x-linked disorder occuring in one out
of every 3,500 liveborn males. By age nine, most of the boys are unable to walk
and many die within a few years because of cardiac failure. Becker muscular
dystrophy (BMD) on the other hand is caused by frameshift mutations only 16
% of the time, where the most common mutations are in-frame deletions (Pestronk,
2002). BMD is much less severe than DMD, where DMD dystrophin is not made
at all or lacks the ß-dystroglycan binding domain but BMD patients have
partially functional dystrophin proteins. Diagnosis of muscular dystrophy in
a developing fetus is possible through chorionic villus sampling (CVS) or amniotic
fluid (AF) cell cultures to test for the presence of the normal dystrophin protein.
Current research to discover a possible cure is using the homologous gene in
mice, mdx mouse, to use gene therapy in rescuing dystrophin (Green,
2001).
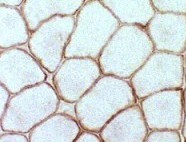
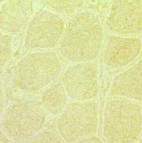
| Figure 4. Immunostaining of dystrophin in normal cells (left) and DMD cells (right). Dystrophin is found around each muscle fiber in normal cells, but is absent or present in very small quantities in DMD cells. Permission pending from author, see source. |
References
Green, E. (2001). “The Dystrophin Story: NMD leads to new understanding of Muscular Dystrophy.” (Site visited on 25 Feb. 2003: http://compbio.berkeley.edu/people/ed/rust/Dystrophin.html)
López, L., Fickett, J. (1997). “Catalogue of Regulatory Elements.” 16 Mar. 1997. (Site visited on 25 Feb. 2003: http://www.cbil.upenn.edu/MTIR/dys-when.html)
Malcolm, S. (2001) “Structure and Function of the Dystrophin Gene.” 8 Aug. 2001. (Site visited on 25 Feb. 2003: http://www.ich.ucl.ac.uk/cmgs/dmdgene99.htm)
Nudel, U., Yaffe, D., Fuchs, O., Leibovitz, S., Neuman, S. (2001). “The Duchenne Muscular Dystrophy Gene: Structure, Evolution, Expression and Function of Products.” (Site visited on 27 Feb. 2003: http://www.weizmann.ac.il/Biology/open_day/book/uri_nudel.pdf)
Pestronk, A. (2002). “Neuromuscular Disease Center at Washington University: Dystrophinopathies.” 9 Feb. 2003. (Site visited on 25 Feb. 2003: http://www.neuro.wustl.edu/neuromuscular/musdist/dmd.html#dp)
Sugita, S., Saito, F., Tang, J., Satz, J., Campbell, K., Sudhof, TC. (2001). “A Stoichiometric Complex of Neurexins and Dystroglycan in Brain.” Journal of Cell Biology. 23, 435-45 (Site visited on 27 Feb. 2003: http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&uid=11470830&Dopt=r)
![]()
![]()