
This web page was produced as an assignment for an undergraduate course at Davidson College
PrP Orthologs
Images by Clip Art in Microsoft Word
Comparing Prion Orthologs
The prion protein has been isolated from a range of species (mostly vertebrates) and is found only in eukaryotes. PrP orthologs have an average of 257 residues and approximately 17 phenotypic alleles (Mouse Genome Informatics, 2005). The most studied orthologs include those of the primates, including humans and chimpanzees, rodents, including rats and mice, and ungulates such as domesticated pigs, sheep, goats, and cows, among others (Sanger Institute- Ensembl Protein Report, 2005). The analysis of PrP orthologs is of particular importance as spongiform encephalopathies (SEs), resulting from altered forms of normal PrP, can be transmitted across species. Transmission is facilitated by high sequence homology among prion orthologs of diverse species. Here, I compare the amino acid sequences of 6 prion orthologs: Homo sapien (human), Mus musculus (mouse), Canis familiaris (dog), Ovis aries (sheep), Sus scrofa (pig), and Bos taurus (cow) (Figure 1) ( Sanger Institute- Ensembl Protein Report, 2005; National Center for Biotechnology Information- Entrez Protein, 2005 ).
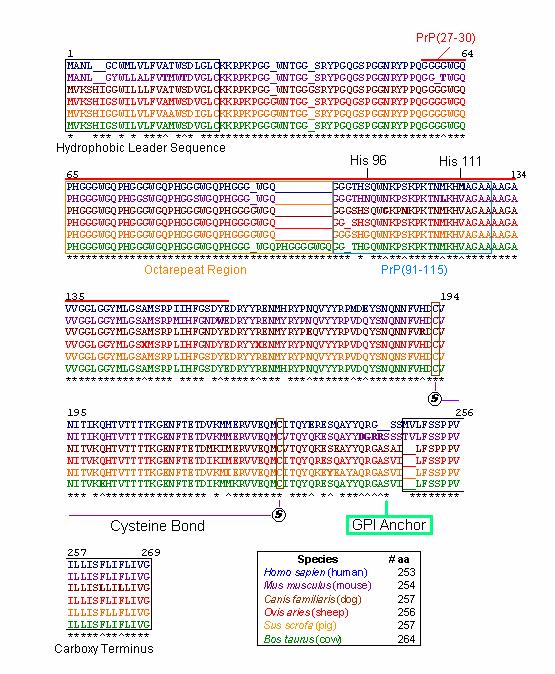
Figure1: Amino acid sequences of selected PrP protein orthologs. Illustrated in blue, purple, brown, red, yellow, and green are the PrP amino acid sequences of Homo sapien (human), Mus musculus (mouse), Canis familiaris (dog), Ovis aries (sheep), Sus scrofa (pig), and Bos taurus (cow), respectively . Asterisks (*) represent the same amino acid for all orthologs at that position (for all amino acids that exist at that position). Up arrows (^) represent the same amino acid for all orthologs with the exception of one species which is illustrated in bold. Underlines (_) represent artificial spaces within the sequence that allow the orthologs to be aligned properly. Black numbers indicate residue positions. For the purposes of alignment, artificial spaces are counted as residues. The hydrophobic leader and carboxy terminus sequences are boxed in black. PrP(27-30), the truncated version of PrP Sc, is lined in red. The octarepeat and PrP(91-115) regions are boxed in orange and blue, respectively. Positions of HIS 96 and HIS 111, the cysteine bond, and the GPI anchor are also indicated. A legend denotes the number of amino acids in each of the prion orthologs.
Overall, among PrP orthologs there is a high degree of sequence homology, with more than 85% of amino acids shared across species ( McCusker et al., 2005) . Indeed, an examination of the six orthologs illustrated above indicates the most highly conserved regions in the protein. Those regions that are most highly conserved are so because they are critical to normal PrP function. Comparisons of consensus sequences among diverse species allow us to construct phylogenetic trees based on the evolutionary development of PrP orthologs over time and among different species.
The first 24 residues of the prion protein constitute the hydrophobic leader sequence, indicated by a black box (Figure 2). This sequence is one of the least conserved sequences among prion orthologs, with only 58% homology between species. This low sequence homology is likely due to the fact that this region plays only a minor role in prion function and is removed as the prion protein prepares to anchor to the cell surface ( McCusker et al., 2005) . This suggests that the protein may still be able to function despite spontaneous mutations and deletions within this region.

Figure 2: PrP Hydrophobic Leader sequences of the 6 orthologs . Illustrated in blue, purple, brown, red, yellow, and green are the PrP amino acid sequences of Homo sapien (human), Mus musculus (mouse), Canis familiaris (dog), Ovis aries (sheep), Sus scrofa (pig), and Bos taurus (cow), respectively.
The hydrophobic leader sequence also generates information about the prion protein’s phylogenetic tree. A branch in the tree is apparent between species whose sequences begin with ‘MANL’ ( mice & humans) and those species whose sequences begin with ‘MVKS’ ( un gula tes & dogs) (Figure 2). Other branches are suggested by residue 8, which may consist of one of four different amino acids; Cysteine ( human), Tyrosine ( mouse), Glycine ( dog & pig), and Serine ( sheep & cow). These observations are consistent with the phylogenetic tree constructed by Krakauer et al. (Figures 2 & 3).
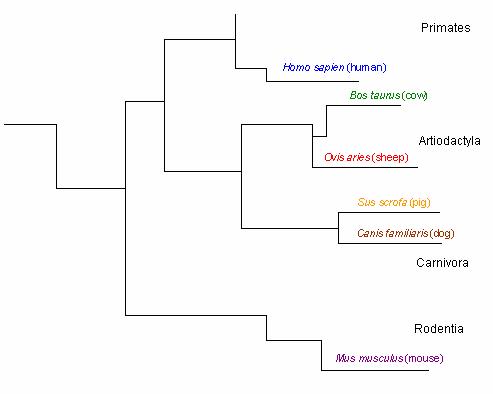
Figure 3: Phylogenetic tree of the prion protein based on ortholog residues . Figure adapted from Krakauer et al. Only the branches leading to the selected 6 orthologs are illustrated. Black letters represent orders.
Recall that when PrP Sc enters a normal cell, it converts PrP C into more PrP Sc. In response, the cell’s protective proteinases attack and attempt to destroy the foreign protein. PrP Sc, however, is resistant to proteinases and is merely truncated to a version of PrP Sc called PrP(27-30) (Zanusso et al., 2004) . This region is indicated by a bold red line (Figure 1). PrP(27-30) includes the octarepeat region, PrP(91-115), HIS 96 and HIS 111 regions.
Of the 6 orthologs studied, 100% sequence homology was found within the octarepeat region (Figure 4). High sequence homology in this region is imperative because the octarepeat region is critical to the function of PrP C in copper homeostasis. Each octarepeat ( PHGGGWGQ) binds one copper molecule after activation by the copper dependent hinges HIS 96 and HIS 111 (Zanusso et al., 2004) . All of the species have 4 octarepeats with the exception of the cow, which has 5 octarepeats. Although the addition of octarepeats alone are known to cause spongiform encephalopathies in humans and other species, the additional octarepeat in Bos taurus is not thought to be linked to bovine spongiform encephalopathy (BSE) ( McCusker et al., 2005).
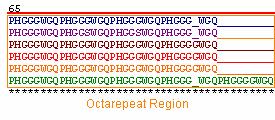
Figure 4: PrP Octarepeat regions of the 6 orthologs . Illustrated in blue, purple, brown, red, yellow, and green are the PrP amino acid sequences of Homo sapien (human), Mus musculus (mouse), Canis familiaris (dog), Ovis aries (sheep), Sus scrofa (pig), and Bos taurus (cow), respectively.
PrP(91-115), including HIS 96 and HIS 111 regions, is thought to be the domain that misfolds to yield the deadly PrP Sc conformation of the prion protein (Jones et al., 2004). This region is slightly less conserved among orthologs, even at HIS 96 and HIS 111 residues (Figure 5). This observation suggests that perhaps HIS 96 and HIS 111 regions are not as critical to protein function or that these copper ‘hinges’ are less sequence specific. PrP(91-115) may be more susceptible to misfolding because mutations are more likely to persist in regions that are less critical to protein function. It is important to note that the residues numbered in this region (i.e. HIS ‘96’) are based on the mice residues because this is the species in which these regions were first identified (Jones et al., 2004).
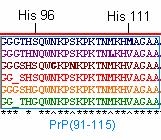
Figure 5: PrP(91-115) of the 6 orthologs . Illustrated in blue, purple, brown, red, yellow, and green are the PrP amino acid sequences of Homo sapien (human), Mus musculus (mouse), Canis familiaris (dog), Ovis aries (sheep), Sus scrofa (pig), and Bos taurus (cow), respectively.
Interestingly enough, the truncation of PrP to PrP(27-30) results in the loss of cysteine residues which anchor the normally intact cysteine bonds in normal PrP (Figure 1) (Zanusso et al., 2004) . Recall that in the formation of PrP Sc, this cysteine bond breaks apart, destabilizing the protein and exposing the unbound sulfurs on the cysteine complex towards the outside of the protein ( McCusker et al., 2005) . Therefore, it can be inferred that the intact cysteine bond is critical to protein function as illustrated by the 100% homology at cysteine anchor sites observed among the 6 orthologs (Figure 1). In addition, these two cysteine residues, C193 and C228, are the only cysteines present in the mature prion protein (excluding the hydrophobic leader sequence ).
The attachment site of prion’s glycosyl phosphatidyl inositol (GPI) anchor is located near the protein’s C terminus at residue 245 (Figure 6) (Jones et al., 2004). Recall that PrP is a glycoprotein tethered to the cell surface via this GPI anchor (Jones et al., 2004). Thus, we can infer that this GPI is critical to PrP function as suggested by the 100% homology among orthologs.

Figure 6: Location of Prion GPI anchor site for the 6 orthologs . Illustrated in blue, purple, brown, red, yellow, and green are the PrP amino acid sequences of Homo sapien (human), Mus musculus (mouse), Canis familiaris (dog), Ovis aries (sheep), Sus scrofa (pig), and Bos taurus (cow), respectively.
Human Mutations Leading to Spongiform Encephalopathy
Transmission of PrP Sc from one organism or species to another is just one of the ways through which an individual can acquire a fatal spongiform encephalopathy. Prion diseases can also occur in inherited or sporadic forms (Collinge et al., 1994). These forms have been studied most extensively in humans who can inherit or sporadically acquire Creutzfeldt-Jakob disease (CJD), Gerstmann-Straussler-Scheinker disease (GSS), and Familial Fatal Insomnia (FFI) (Collinge et al., 1994 ; Montagna et al., 2003). Although at least 18 mutations within the PrP gene leading to these prion diseases have been identified, only a few of them will be described below (Collinge et al., 1994).
Like many prion diseases, CJD can be inherited or may be caused by spontaneous mutations in one or both alleles of the PrP gene (Collinge et al., 1994). About 15% of CJD cases are inherited (the disease is autosomal dominant), while approximately 85% of cases are caused by spontaneous mutations in the genome (Collinge et al., 1994). Symptoms of CJD include progressive dementia and cerebral ataxia. An infected individual generally dies within three months of onset (Collinge et al., 1994).
A study by Wadsworth et al. (2004) found that the generation of the CJD phenotype in mice required that methionine be present at residue 129 in PrP. In addition, they found that the expression of valine at position 129 instead of methionine resulted in a barrier to the transmission of BSE (Wadsworth et al. , 2004). Interestingly enough, M129, or methionine at residue 129, is 100% conserved across the 6 orthologs studied (Figure 7).
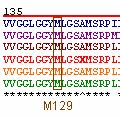
Figure 7: Position M129 of prion orthologs . Illustrated in blue, purple, brown, red, yellow, and green are the PrP amino acid sequences of Homo sapien (human), Mus musculus (mouse), Canis familiaris (dog), Ovis aries (sheep), Sus scrofa (pig), and Bos taurus (cow), respectively. All selected orthologs have methionine present at residue 129.
Scientists speculate that this M/V polymorphism at residue 129 is important in assessing an individual’s genetic susceptibility to prion and prion-like diseases (Collinge et al., 1994) . In fact, a related study by Riemenschneider et al. (2004) found that patients homozygous for methionine at residue 129 were at increased risk of Alzheimer’s disease, with a decreased age at onset.
A study on Kuru, a prion disease common among the Fore linguistic group of Papua New Guinea resulting from ritualistic cannibalism, found that heterozygosity at residue 129 (M/V) conferred resistance to prion diseases (Mead et al., 2003) . Their theory was supported by findings that elderly members of the Fore population had higher frequencies of heterozygosity than younger generations. These data are consistent with the aforementioned study by Wadsworth et al. (2004) .
A study by Mishra et al. (2003) examined point mutations common in CJD patients. They most common substitutions isolated were those valine (V) for isoleucine (I) at human residue 203 and glutamic acid (E) for glutamine (Q) at residue 211 (Figure 8) (Mishra et al. , 2003).

Figure 8: Mutations Associated with Human PrP . Mutations leading to Creutzfeldt-Jakob disease (CJD), Gerstmann-Straussler-Scheinker disease (GSS), and Familial Fatal Insomnia (FFI) are boxed in blue, red, and green, respectively. M129 polymorphisms are associated with all three prion diseases. Black arrows and the letters under them represent mutant residue substitutions. The 8 octarepeat inserts (PDGGGWGQ) are associated with Huntington’s Disease (HD).
The symptoms of GSS are similar to those of CJD except that the duration of time over which the disease takes its course is longer (Collinge et al., 1994). GSS is characterized by the buildup of extensive amyloid deposits within the brain (Kundu et al., 2003). Like CJD, GSS follows an autosomal dominant pattern of inheritance and is thought to be associated with homozygosity of methionine at residue 129 (Kundu et al., 2003).
In 1991, Hsiao et al. found that GSS is also caused by a missense mutation at codon 102 of the open reading frame of PrP. It is thought that in mutated PrP proline (P) is substituted by leucine (L) (Figure 8). To prove their theory, Hsiao et al. (1991) demonstrated that transgenic mice homozygous for leucine at this residue spontaneously developed SE. Indeed, even among orthologs, proline at this position is highly conserved (Figure 9).

Figure 9: Position P102 of prion orthologs . Illustrated in blue, purple, brown, red, yellow, and green are the PrP amino acid sequences of Homo sapien (human), Mus musculus (mouse), Canis familiaris (dog), Ovis aries (sheep), Sus scrofa (pig), and Bos taurus (cow), respectively.
In a second study by Mishra et al. (2003), GSS was found to be the result of the substitution of glutamine (Q) for proline (P) at human residue 212. Another study found that GSS resulted from the substitution of tyrosine (Y) for a stop codon at residue 145 (Figure 8) (Kundu et al., 2003).
FFI is characterized by disrupted sleep, hyperactivity, hypometabolism, and motor abnormalities (Montagna et al., 2003). FFI is thought to be the result of the combined effect of the substitution of aspartic acid (D) for asparagine (N) at human residue 178 and a polymorphism, V for M, at codon 129 (Figure 8) (Montagna et al., 2003). CJD is also thought to be caused by a similar mutation (D178N) at that residue with an altered three-dimensional form of the mutated PrP protein (Montagna et al., 2003).
PrP mutations and Huntington’s Disease?
A study by Moore et al. (2001) revealed that Huntington’s disease (HD) may be linked to mutations within the PrP gene on human chromosome 20. It was found that individuals with HD were also heterozygous for a 192-nucleotide insertion within the octarepeat region of PrP (Moore et al. , 2001). Instead of the typical 4 octarepeats present in the human ortholog, HD patients had an additional eight octapeptide repeats (Figure 8) (Moore et al. , 2001). As aforementioned, the octarepeat region is critical to the function of PrP C in copper homeostasis as each octarepeat ( PHGGGWGQ) binds one Cu 2+ molecule (Jones et al. , 2004) . Thus, insertions within this region likely induce HD and, indeed, Moore et al. (2001) found that PrP insertions provoked early onset of HD with symptoms remarkably similar to those displayed by CJD patients.
PrP and the Human Immunodeficiency Virus?
A recent study by Leblanc et al. (2004) found that PrP C expressed a nucleic acid binding and chaperoning properties similar to those of a nucleocapsid protein of human immunodeficiency virus type 1 (HIV-1). Nucleocapsids are proteins that coat the capsid of a virus containing the virus’ nucleic acid genome. Interestingly enough, Leblanc et al. (2004) found that an increase in PrP C expression correlated with a three to four fold decrease in HIV-1 infectivity and an eightfold decrease in virus production. Their study also demonstrated the importance of the aforementioned GPI anchor as PrP C mutants without this anchor did not confer decreases in HIV-1 infectivity and virus production ( Leblanc et al. , 2004) .
Summary of PrP Mutations
Disease |
Residue |
Normal Residue |
Mutant Residue |
CJD/GSS/FFI |
129 |
M |
M/V polymorphisms |
CJD |
203 |
V |
I |
CJD |
211 |
E |
Q |
GSS |
102 |
P |
L |
GSS |
212 |
Q |
P |
GSS |
145 |
Y |
stop |
FFI |
178 |
D |
N |
HD |
octarepeat region |
4 octarepeats |
12 octarepeats |
Links
http://www.cjdsurveillance.com/ National Prion Disease Pathology Surveillance Center
http://www.mad-cow.org/ The Official Mad Cow Disease Homepage
Works Cited
Collinge, J. and M. Palmer. 1994. “Molecular genetics of human prion diseases.” Philosophical Transactions of the Royal Society of London. 343: 371-8.
Hsiao, K., and S.B. Prusiner. 1991. “Molecular genetics and transgenic model of Gertsmann-Straussler-Scheinker disease.” Alzheimer Disease and Associated Disorders. 5(3):155-62. <http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=1685324> Accessed 2005 8 Mar.
Jones, C. E., Abdelraheim, S., Brown, D., and J.H. Viles. 2004. “Preferential Cu 2+ Coordination by His 96 and His 111 Induces B-Sheet Formation in the Unstructured Amyloidogenic Region of the Prion Protein.” Journal of Biological Chemistry. 279(31): 32018-32027.
Kundu, B., Maiti, N.R., Jones, E.M., Surewicz, K.A., Vanik, D.L., Surewicz, W.K. 2003. “Nucleation-dependent conformational conversion of the Y145Stop variant of human prion protein: structural clues for prion propagation.” Proceedings of the National Academy of Science U.S.A . 100(21):12069-74. < http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=14519851> Accessed 2005 8 Mar.
Leblanc, P., Baas, D., and J.L. Darlix. 2004. “ Analysis of the interactions between HIV-1 and the cellular prion protein in a human cell line.” Journal of Molecular Biology. 337(4):1035-51. <http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15033368> Accessed 2005 7 Mar.
McCusker, R., Novakofski, J., and S. Brewer. 2005. Bovine Spongiform Encephalitis Information Online. Available <http://w3.aces.uiuc.edu/AnSci/BSE/> Accessed March 2, 2005.
Mead, S., Stumpf, M.P., Whitfield, J., Beck, J.A., Poulter, M., Campbell, T., Uphill, J.B., Goldstein, D., Alpers, M., Fisher, E.M., and J. Collinge. 2003. “Balancing selection at the prion protein gene consistent with prehistoric kurulike epidemics.” Science. 300(5619):640-3. < http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=12690204> Accessed 2005 8 Mar.
Montagna, P., Gambetti, P., Cortelli P. and E. Lugaresi. 2003. “Familial and sporadic fatal insomnia.” Lancet Neurology. 2(3):167-76. < http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=12849238> Accessed 2005 8 Mar.
Moore, R., Xiang, F., Monaghan, J., Han, D., Zhang, Z., Edström, L., Anvret, M., and S. Prusiner. 2001. “ Huntington Disease Phenocopy Is a Familial Prion Disease.” American Journal of Human Genetics. 69:1385-1388. < http://www.journals.uchicago.edu/AJHG/journal/issues/v69n6/013165/013165.html> Accessed 2005 7 Mar.
Mouse Genome Informatics, 2005. Sequence Detail: Prnp. Available < http://www.informatics.jax.org/javawi2/servlet/WIFetch?page=sequenceDetail&id=NM_011170> Accessed February 10, 2005.
National Center for Biotechnology Information, 2005. Entrez Protein: Bos taurus Prnp. Available <http://www.ncbi.nlm.nih.gov/entrez/viewer.fcgi?db=protein&val=41386735> Accessed March 3, 2005.
National Center for Biotechnology Information, 2005. Entrez Protein: Mus musculus Prnp. Available <http://www.ncbi.nlm.nih.gov/entrez/viewer.fcgi?db=protein&val=13879449> Accessed March 3, 2005.
National Center for Biotechnology Information, 2005. Entrez Protein: Ovis aries Prnp . Available <http://www.ncbi.nlm.nih.gov/entrez/viewer.fcgi?db=protein&val=59596166> Accessed March 3, 2005.
National Center for Biotechnology Information, 2005. Entrez Protein: Sus scrofa Prnp. Available <http://www.ncbi.nlm.nih.gov/entrez/viewer.fcgi?db=protein&val=56711358 > Accessed March 3, 2005.
Riemenschneider, M., Klopp, N., Xiang, W., Wagenpfeil, S., Vollmert, C., Muller, U., Forstl, H., Illig, T., Kretzschmar, H., and A. Kurz. 2004. "Prion protein codon 129 polymorphism and risk of Alzheimer disease." Neurology. 63: 364-366 <http://www.neurology.org/cgi/content/abstract/63/2/364> Accessed 2005 7 Mar.
Sanger Institute, 2005. Ensembl Human Genome Server: Canis familiaris PNRP. Available <http://www.ensembl.org/Canis_familiaris/protview?peptide=ENSCAFP00000009107&db=core> Accessed March 5, 2005.
Sanger Institute, 2005. Ensembl Human Genome Server: Homo sapien PNRP. Available <http://www.ensembl.org/Homo_sapiens/protview?peptide=ENSP00000306540&db=core> Accessed March 3, 2005.
Sanger Institute, 2005. Ensembl Human Genome Server: PNRP. Available < http://www.ensembl.org/Homo_sapiens/geneview?gene=OTTHUMG00000031786&db=vega> Accessed March 3, 2005.
Wadsworth , J.D., Asante, E.A., Desbruslais, M., Linehan, J.M., Joiner, S., Gowland, I., Welch, J., Stone, L., Lloyd, S.E., Hill, A.F., Brandner, S. and J. Collinge. 2004. “Human prion protein with valine 129 prevents expression of variant CJD phenotype.” Science. 306(5702):1793-6. < http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15539564> Accessed 2005 7 Mar.
Zanusso, G., Farinazzo, A., Prelli, F., Fiorini, M., Gelati, M., Ferrari, S., Righetti, P., Rizzuto, N., Frangione, B., and S. Monaco . 2004. “Identification of Distinct N-Terminal Truncated Forms of Prion Protein in Different Creutzfeldt-Jakob disease Subtypes.” Journal of Biological Chemistry. 279(37): 38936-38942.
Questions or Comments? Please e-mail Katie Winter