X-Linked Agammaglobulinemia
X-Linked Agammaglobulinemia (XLA) was the first immunodeficiency
disease to be described. It was first characterized by Ogden C. Bruton
in a 1952 case study of an eight year old boy (Bruton, 1952). Bruton
describes the boy as having chronic infections with a variety of pathogens
over a four year period, each of which was successfully treated with penicillin
or a sulfa drug of some variety. Since the majority of his infections
had been caused by pneumococcus, Bruton attempted to vaccinate him against
this pathogen. When it was discovered that no gamma globulin was
produced by the vaccination, the boy was tested to determine whether he
could produce antibodies to any antigen. It was soon proven that
the boy could produce virtually no gamma globulin in response to any pathogen.
Bruton therefore named the condition agammaglobulinemia (Bruton, 1952).
The condition was soon observed to only occur in males and was therefore
determined to be X-linked. The disease is now commonly known as Bruton's
X-Linked Agammaglobulinemia.
Historical Background
When Bruton first described agammaglobulinemia in 1952, he suggested that there were two possible causes for the condition; the first being "a congenital dysfunction in the mechanism of gamma globulin production" and the second being "an acquired dysfunction in this mechanism" (Bruton, 1952). Bruton felt that the disease was acquired since the boy in his case study exhibited no symptoms until after he reached four years of age. The observation that XLA was X-linked ruled out Bruton's second hypothesis, indicating that the cause of the disease was genetic. The cause of the condition was further narrowed with the discovery that patients with XLA lack mature B lymphocytes (Geha et al., 1974). Further research indicated that in addition to the absence of mature B-cells in the periphery, patients with XLA also showed a large decrease in the ratio of pre B-cells to pro B-cells in the bone marrow (Camapana, 1990). This result indicates that the disease is caused by something arresting the development of B-cells between the pro B-cell and pre B-cell stages. A major step toward the understanding of the cause of XLA was taken in 1993 with the independent isolation of the gene which, when mutated, causes XLA (Vetrie et al., 1993; Tsukada et al., 1993). As expected, the gene was located on the X-chromosome. The gene was found to code for a protein kinase which had not yet been observed (Vetrie et al., 1993). This new protein kinase was named Bruton's Tyrosine Kinase (Btk) because of its role in XLA.
Current Understanding
Btk is believed to play a role in a kinase signaling cascade
that promotes the rearrangement of the light chain in pre B-cells after
the pre B-cell receptor is displayed on the B-cell's surface, although
the exact role of Btk in this pathway is not yet known (Janeway et al.,
1999). Btk is known to consist of five distinct domains: Pleckstrin
homology (PH), Tec homology (TH), Src homology 3 (SH3), Src homology 2
(SH2), and the catalytic kinase domain (Ki) (Vihinen et al., 1999).
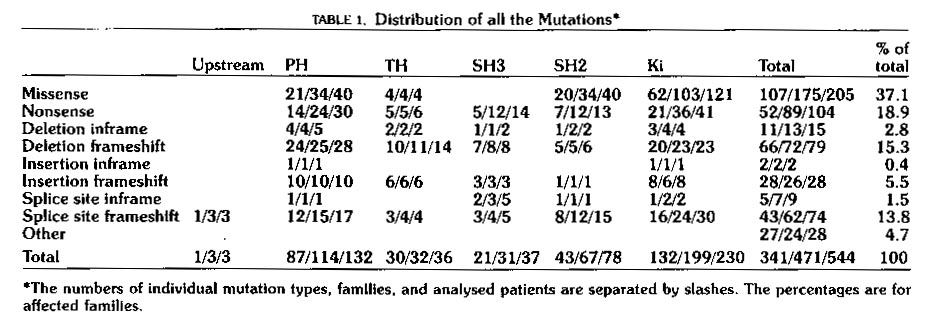
Mutations causing XLA are distributed over all five domains as is shown
in Table 1 below.
Table 1: Distribution of mutations in Btk known to cause XLA.
The table shows that mutations are distributed over all five domains.
Mutations of all types in any of the five domains seem to be able to lead
to XLA. This table is reproduced as printed in Vihinen et
al,
1999.. Permission to use this image has been requested, and if
denied, the table will be removed from this page.
As is shown in Table 1 above, many different types of
mutations in any of the five domains can lead to the XLA phenotype.
These data represent those XLA causing mutations of Btk which were known
at the date of publication of Vihinen's paper. A constantly updated
list, known as BTKbase is maintained at http://www.uta.fi/imt/bioinfo/BTKbase/.
Case studies have shown that mutations in sites that are generally conserved
from person to person lead to severe (classical) XLA in which virtually
no B-cell development is observed, while mutations in sites which are somewhat
more variable lead to a milder form of the disease (Vihinen et al.,
1999). The fact that mutations occurring in any of the five domains
can cause XLA suggests that each of Btk's domains plays a vital role in
B-cell development. This hypothesis is supported by many recent studies
of the activity of the domains of Btk in vitro.
PH Domain
In order to become fully active, Btk must bind to the
plasma membrane and dimerize, allowing trans-phosphorylation of the SH3
domain of each monomer. The PH domain of Btk is known to be capable
of binding phospholipid head groups in vivo (Baraldi et al.,
1999). XLA causing mutations in the PH domain have been divided into
two groups: those that are large enough to disrupt the folding of
the domain and those that alter the domain's affinity for phospholipid
head groups (Baraldi et al., 1999). Clearly, both types of
mutations would interfere with the activation of Btk and prohibit it from
acting on other kinase molecules in the signaling cascade.
TH and SH3 Domains
As stated above, activation of Btk requires dimerization
and trans-phosphorylation of the SH3 domains. Recent research in
vitro has shown that the SH3 domain is capable of binding strongly
with the proline- rich TH domain, making both domains unavailable for interaction
with other proteins (Patel et al., 1997; Desiderio et al.,
1997). It is likely that this intramolecular binding plays a role
in the regulation of the activity of Btk. It has been shown that
mutations in either the TH or SH3 domains can lead to a decrease in the
avidity of binding between the two domains (Patel et al., 1997).
This change in avidity could interfere with the ability of Btk to respond
to and pass on signals in a kinase cascade and curtail B-cell development.
SH2 and Ki Domains
Important roles of SH2 and kinase domains are known to
be binding to phosphorylated tyrosine residues and phosphorylating tyrosine
residues, respectively. Mutations in these domains of Btk could easily
interfere with Btk's ability to receive signals by binding to phosphorylated
tyrosine residues with its SH2 domain, or to pass signals on by catalyzing
the phosphorylation of another molecule with its kinase domain. The
fact that mutations in these domains do interfere with Btk activity are
further evidence that Btk is a member of a kinase cascade which is critical
to B-cell development (Vihinen, 1999).
Additional Functions
As research into XLA continues, more of Btk's roles are
likely to be discovered. For example, recent work has shown links
between mutations in Btk and the activity and location of the nuclear transcription
factor TFII-I (Novina et al., 1999). As more functions are
discovered, it will continue to become clear that the complexity of intracellular
communication requires each protein to perform multiple roles, leading
to many possibilities for mutation. The same phenotype can often
be caused by any one of these mutations, making localization of the cause
of disease difficult and making the use of new diagnostic and treatment
techniques extremely important.
Symptoms and Methods of Diagnosis
As described by Bruton, males who are affected by XLA are extremely vulnerable to bacterial infection and are faced with recurrent infections. These children are often susceptible to infection by a normally harmless pathogen given as a vaccination. Such cases have been observed with the polio vaccine (Wright et al., 1977). Diagnosis of XLA is made at an early age by observation of a lack of immunoglobin in the blood stream and the specific mutation which is causing the disease can be determined by genetic analysis. A current clinical synopsis of the effects of XLA is available through OMIM (Online Mendelian Inheritance in Man).
Females who are carriers of the disease show no symptoms because all of their mature B-cells exhibit exclusive use of their normal X-chromosome. This non-random X-inactivation is confined to the carrier's B-cells, indicating that the mutation does not affect cells of other lineages, including T-cells and macrophages (Conley et al., 1986). Thus, carriers of XLA can easily be identified by the presence of non-random X-inactivation.
The first successful treatment of XLA was initiated by Bruton in 1952. He treated his young patient with monthly injections of gamma globulin, with exceptional results. The boy, who had suffered nineteen cases of sepsis over the past several years, displayed no sepsis during the fourteen months in which he was receiving injections (Bruton, 1952). This method remained the standard for treatment of XLA until the development of intravenous immunoglobin (IVIG) in 1986 (Lee et al., 1997). The IVIG method was preferred over intramuscular injections because it allowed for the injection of larger amounts of immunoglobin much less painfully. A long term study of the effectiveness of IVIG indicated that it was successful in preventing and treating bacterial infections; however, its effectiveness against viral infections was much more limited (Quartier et al., 1999).
The most recent research in this field has yielded encouraging
results on the possibility of the use of transgenes to restore Btk activity.
Studies in mice with the similar X-linked immunodeficiency (XID) have shown
that the XID mutation has no dominant negative effect: the insertion
of a Btk transgene was able to restore antiviral TI-2 antibody responses
in these mice even though the gene was inserted into a different position
than in the wild type mice (Pinschewer et al., 1999). This
research is still far from providing an option for human patients suffering
from XLA, but it does provide a glimpse of what will likely become the
preferred method of treatment of XLA and similar genetic diseases.
Baraldi, E., K.D. Carugo, M. Hyvönen, P.L. Surdo,
A.M. Riley, B.V. Potter, R. O'Brien, J.E. Ladbury, and M. Saraste. 1999.
Structure.
7, 449-460.
Bruton, O.C. 1952. Pediatrics.
9,
722-728.
Campana, D., J. Farrant, N. Inamdar, A.D. Webster, G.
Janossy. 1990. J. Immunol. 145,
1675.
Conley, M.E., P. Brown, A.R. Pickard, R.G. Buckley, D.S.
Miller, W.H. Rasking, J.W. Singer, and P.J. Fialkow. 1986.
New
England Journal of Medicine. 315, 564-567.
Desiderio, S. 1997. Current Opinion
in Immunology. 9, 534-540.
Fuleihan, R., R. Narayanaswamy, and R.,S. Geha. 1995.
Advances
in Immunology. 60, 37-56.
Geha, R.S., F.S. Rosen, and E. Merler. 1974.
J.
Clin. Invest. 52, 1726.
Lee, M.L., R.P. Gale, and P.L. Yap. 1997.
Annu.
Rev. Med. 48, 93-102.
Novina, C.D., S. Kumar, U. Bajpai, V. Cheriyath, K. Zhang,
S. Pillai, H.H. Wortis, and A.L. Roy. 1999. Molecular
and Cellular Biology. 19, 5014-5024.
OMIM: Online Mendelian Inheritance in Man.
2000. Clinial Synopsis for OMIM ENTRY 300310. <http://www3.ncbi.nlm.nih.gov/htbin-post/Omim/dispmim?300310.cs>
Accessed 2000 Apr 14.
Patel, H.V., S. Tzeng, C. Liao, S. Chen, and J. Cheng.
1997.
Proteins:
Structure, Function, and Genetics.
29, 545-552.
Pinschewer, D.D., A.F. Ochsenbein, A.B. Satterthwaite,
O.N. Witte, H. Hengartner, and R.M. Zinkernagel. 1999. European
Journal of Immunology. 29, 2981-2987.
Quartier, P., M. Debré, J. DeBlic, R. de Sauverzac,
N. Sayegh, N. Jabado, E. Haddad, S. Blanche, J. Casanova, C.I. Smith, F.
Le Deist, G. de Saint Basil, and A. Fischer. 1999. The
Journal of Pediatrics. 134, 589-596.
Tsukada, S., D.C. Saffran, D.J. Rawlings, O. Parolini,
R.C. Allen, I. Klisak, R.S. Sparkes, H. Kubagawa, T. Mohandas, S. Quan,
J.W. Belmont, M.D. Cooper, M.E. Conley, and O.N. Witte. 1993.
Cell.
72,
279-290.
Vetrie, D., I. Vorechovsky, P. Sideras, J. Holland, A.
Davies, F. Flinter, L. Hammarström, C. Kinnon, R. Levinsky, M. Bobrow,
C.I. Smith, and D.R. Brently. 1993. Nature. 361,
226-233.
Vihinen, M., S. Kwan, H.D. Ochs, I. Resnick, J. Väliaho,
M.E. Conley, and C.I. Smith. 1999. Human Mutation.
13,
280-285.
Vihinen, M. and C.I. Smith. 2000. BTKbase:
Mutation registry for X-linked agammaglobulinemia. <http://www.uta.fi/imt/bioinfo/BTKbase/>
Accessed 2000 Apr 13.
Return to Bob Magnussen's Immunology Home Page
Return to Immunology Main Page
![]()
Send comments, questions, and suggestions to: bomagnussen@davidson.edu