
Nature Genetics 7: 497-501, 1994.
article
B. Bardoni1, E. Zanaria1, S. Guioli1, G. Floridia1, K. C. Worley2, G. Tonini3, E. Ferrante4, G. Chiumello5, E. R. B. McCabe2, M. Fraccaro1, O. Zuffardi1 & G. Camerinol1,6
1Biobgia Generale e Genctica
Medica, Universita' di Pavia, via Forlanini 14, 27100 Pavia, Italy
2Department of Molecular and
Human Genetics1 Baylor College of Medicine, 1 Baylor Plaza, 77030
Houston, Texas, USA
3Istituto per l'Infanzia, via
dell' Istria 65/1, 34100 Trieste, Italy
4Istituto di Clinica Pediatrica,
Universita' La Sapienza, viale Regina Elena 324, 00161 Roma, Italy
5Clinica Pediatrica III, Ospedale
S. Raffaelle, via Olgettina 60, 20132 Milano, Italy
6Istituto di Istologia ed Embriologia,
Universita' di Sassari, via S. Pietro 43/B, Sassari, Italy
Male to female sex reversal has been observed in individuals with duplications of the short arm of the X chromosome. Here we demonstrate that sex reversal results from the presence of two active copies of an Xp locus rather than from its rearrangement and that alterations at this locus constitute one of the causes of sex reversal in individuals with a normal 46,XY karyotype. We have named this locus DSS (Dosage Sensitive Sex reversal) and localized it to a 160 kilobase region of chromosome Xp21, adjacent to the adrenal hypoplasia congenita locus. The identification of male individuals deleted for DSS suggests that this locus is not required for testis differentiation. We propose that DSS has a role in ovarian development and/or functions as a link between ovary and testis formation.
The existence of an X-specific gene involved in human sex determination was first postulated in 1978, with the identification of a family with an apparent X-linked mode of inheritance of 46,XY gonadal dysgenesis1. A number of families with X-linked recessive (or sex-limited autosomal dominant) transmission of the disorder have been reported since then (reviewed in ref. 2). Further evidence for the involvement of an X-specific locus in sex reversal was provided by the identification of sex reversed patients carrying duplications of portions of Xp. Twelve individuals with partial duplications of the short arm of the X chromosome and an intact SRY gene have been described3-10. Among them, eight had female or ambiguous external genitalia3-7,10 and in two cases histological examination of the gonads demonstrated impaired testis formation3,7.
47,XXY individuals have a single active X chromosome and are male while some of the Xp duplication patients- who all carry a single X inactivation centre- are sex reversed. It was therefore assumed that the Xp-duplication sex reversal is due to double dosage of an X-linked gene(s) which is normally subject to X-inactivation.The alternative hypothesis- that all sex reversing Xp duplications have similar breakpoints, disrupting a gene involved in sex determination-could not be exduded since only one patient was extensively examined at the molesular level7.
To distingtush between these hypotheses and to map the X-linked locus responsible for sex reversal, we have analysed 31 male-to-female sex reversed patients, including four with a cytogenetically visible Xp duplication and 27 with a normal XY karyotype. Our results indicate that dosage of a locus on chromosome Xp21 affects sexual differentiation.
Patents
Fig. 1 Extent of the duplications in the eight patients and physical mapping of the DSS critical interval. Eight genetic males with microscopic duplications of Xp were studied by dosage analysis with previously identified DNA markers24,25. Loci were ordered from Xpter-Xp11 .4 and the extent of the critical interval was estimated using available genetic and physical maps24,25. Filled bar, duplicated fragment; open circle, not tested; E.G., extemal genitalia; F. female; A, ambiguous; M, male extemal genitalia.
We have collected eight patients with duplications of portions of the short arm of the X chromosome: three have an intact Y chromosome and a single duplicated X chromosome, while five carry an intact X chromosome and an additional portion of Xp translocated to the Y chromosome (Fig. 1). All eight patients have a grossly intact SRYgene as determined by Southern blot analysis (not shown). Four of the patients were brought up as females, having either female external genitalia (711, BG and PT) or ambiguous external genitalia (RR). Histological examination of the internal genitalia was performed in three of these patients (BG, PT and RR) and confirmed partial gonadal dysgenesis. The other four patients (TM, PD, SR and RA) are phenotypically males (see Fig. 1). All eight patients have a complex phenotype (induding mental retardation and multiple minor malforrnations) that will be described in detail elsewhere.
Sex reversal and Xp21 duplication
The extent of the Xp duplication was studied in all eight patients by Southern blotting dosage analysis with previously identified DNA markers from Xpter-Xp11.4. The duplication breakpoints are different in the four sex reversed patients (Fig. 1), which excludes gene disruption as a cause of sex reversal and strongly supports the hypothesis that double dosage of an Xp locus is responsible for male to female sex reversal. We named this locus DSS (for dosage sensitive sex reversal) . The correlation between the portion of Xp duplicated nd the phenotypic sex of the eight patients defines the minimal region duplicated in all sex reversed patients as the DSS critical interval. This region extends from the distal boundary, located between loci DXS418 and DXS274, to the proximal boundary, between the Duchenne muscular dystrophylocus (DMD) and the DXS319 locus, a region of approximately 20 megabases (Mb) in Xp21.2- p22.1 (Fig. 1).
Refining the DSS critical region
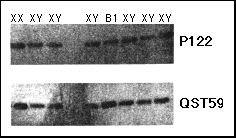
Fig. 2 Southern blotting dosage analysis with probes P122 (DXS418 ) and QST59 (DXS319 ) in patient B1. DNA was digested with EcoRI. The first three lanes on the left are from a normal female and two normal males, respectively; the lanes on the right are from sex reversed 46,XY females.
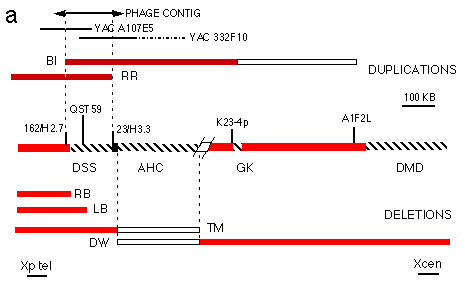
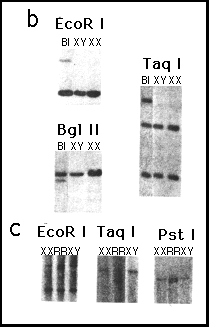
Fig. 3 Physical mapping and overlap cloning at the DSS region.
a. Map of the AHC, GK and DMD region12,
showing the location of the DSS critical interval. Only YACs and
probes described in the text are shown. The extent of the critical
interval was delimited using previously described probes12 and probes obtained from the phage
contig. The extent of the Xp duplication in patients B1 and RR
is shown in the upper part of the Figure, together with the position
of YACs A107E5 and 332F10 and of the phage contig. The proximal
breakpoint of the duplication of B1 was locaiized between probes
A1 F2L and K23-4p. The extent of the deletions (dotted lines)
in previously described AHC patients11,12 is shown in the lower part of the Figure.
Patients LB, RB and TM are affected by AHC, GK and DMD. Patient
DW is affected by AHC. Red bars represent part of the chromosome
that are present, open bars represent regions of uncertainty.YAC
332F10 contains a portion of Xp coligated with a portion of 4q32
(hatched).
b Identification of the distal breakpoint of the duplication of
patient B1 with probe 162/H2.7. Molecular weight of hybridizing
bands were: EcoR I, normal fragment 10 kb, altered size fragment
17 kb; Bgl I normal 10.7 kb, altered 10 kb; Taq I, 2.1 and 1.2
normal, 2.6 altered. XX, normal female; XY normal male.
c, Identification of the X;Y translocation breakpoint in patient
RR using probe 223/H3.3. Molecular weight of hybridizing bands
were: EcoR I, normal fragments 4.5 and 3.5 kb, altered size fragment
4.3 kb; Taq I, normal 5.8 kb, altered 4 kb; Pst I, 2.1 normal,
1.8 altered. XX, normal female; XY normal male.
We speculated that submicroscopic duplications of the DSS locus might be one of the causes of sex reversal in individuals with a normal 46,XY karyotype. The genomes of 27 46,XY sex reversed females were screened for submicroscopic rearrangements with probes mapping to the DSS critical interval. An abnormal, double dosage was detected when the DNA from patient Bl was hybridized with probe QST59 (locus DXS319, mapping within the proximal boundary of the DSS critical interval) (Fig. 2). The region including DXS319 has been cloned previously in yeast artificial chromosomes (YACs) as part of a study of the contiguous gene syndrome of adrenal hypoplasia congenita (AHC), glycerol kinase deficiency (GF and DMD11-13. Probes obtained from the YAC contigs spanning this region (Fig. 3a) and cDNAprobes for DMD were used to define, by Southern blotting dosage analysis, the extent of the duplication in patient B1. The duplication spans less than 1 Mb and does not include DMD (Fig. 3a). We further demonstrated, by FISH analysis with YAC 332F 10, that the duplicated fragments are closely linked in Xp21 (not shown).
These data narrow the DSS critical interval to the region of overlap of the duplications of patients RR and B1. The extent of the interval was defined by dosage analysis of DNA from BI and RR with probes obtained from a contiguous region of approximately 230 kb, subcloned from the YAC contig into a phage vector (Fig. 3a). Probes 162/H2.7 and 223/H3.3 recognize junction fragments of altered size corresponding to the duplication breakpoint of B1 and RR, respectively (Fig. 3b,c). Therefore these probes, located approximately 160 kb apart, provide landmarks for the proximal and distal limits of the position of DSS (Fig. 3a).
The AHC and GK critical intervals were previously characterized by deletion mapping in patients11-13. As the DSS critical intelval maps very close to the AHC critical interval, we used probes obtained from the phage contig to refine the distal limit of the critical interval for AHC Deletion mapping in key patients13,14 indicates that the AHC and DSS critical intervals are adiacent, being separated only by the deletion breakpoint of patient TM, affected by AHC and not deleted for the DSS region (Fig. 3a).
Discussion
Localization of DSS. Duplications of the short arm of the X chromosome have been associated with male to female sex reversal. To understand if the duplication or the rearrangement of an Xp gene is responsible for sex reversal, we have mapped the duplication breakpoint in four sex reversed, Xp-duplicated patients. The finding that both the proximal and the distal breakpoints differ in the four patients strongly suggests that double dosage of an Xp locus, that we have named DSS impairs testis formation in the presence of a functional SRY gene. DSS may be related to the postulated (and unmapped) X-linked gonadal dysgenesis gene, GDXY(MIM 306100)14, responsible for familial cases of 46,XY sex reversal. If this is the case, it may be anticipated that familial GDXY mutations increase DSS activity (see also below).
The DSS locus was initially mapped to a region of approximately 20 Mb of Xp21.2-p22.1 by comparing the extent of the duplications in the four sex reversed patients and in four patients with normal testis differentiation. We speculated that duplication of DSS might be one of the causes of sex reversal in individuals with a normal 46,XY karyotype and screened the genomes of 27 46,XY sex reversed females for submicroscopic chromosomal rearrangements of Xp21.2-p22.1. Our identification of a submicroscopic duplication in a 46,XY sex reversed female places the DSS-critical region in a 160 kb region of Xp21, adjacent to the AHC locus. The DSS and AHC critical intervals are separated by the deletion breakpoint of one patient, affected by AHC and not deleted for the DSS region. It is possible that DSS and AHC correspond to the same locus and that duplications or deletions of the same gene may prevent the correct differentiation of gonads or adrenal glands, respectively.
DSS dosage and gonadal differentiation. The phenotype of the genitalia in the Xp duplicated, sex reversed patients indicates that two doses of DSS are sufficient to disrupt normal testis development in the presence of SRY. These patients show, however, different degrees of gonadal dysgenesis15, ranging from incompletely differentiated testes (patients PT and RR) to the presence of a single streak gonad (patient BG). Patient B1, who carries the smallest of Xp2 1 duplication, has normal female external genitalia and dysgenetic gonads. This indicates that the variability in sexual differentiation is not related to the size of the duplications and in turn suggests that additional Xp genes are not involved in sex reversal. Phenotypic variability is also seen in the sex reversal syndromes associated with mutations in SRY16,17 or in the Wilm's tumour gene18.
The localization of DSS reveals one important feature of the gene. Among the 46,XY patients affected by the complex AHC-GK-DMD deletion syndrome, many are entirely deleted for the DSS critical region (Fig. 3a). All 46,XY patients with deletions in this region have male external genitalia, suggesting that the DSS locus does not play a major role in testis differentiation.
In some of the Xp duplication cases described (cases 711, PT and Bl) the duplication was inherited from the mother4 (data not shown). Similarly, in a few cases of Xp deletion encompassing DSS, the deletion was maternally inherited19. Genotypic females carrying a duplication or a deletion at the DSS locus can thus be fertile. Although it is likely that an increased DSS dosage does not interfere with ovary formation, the effect of its deletion in XX females is difficult to assess, due to X-inactivation. Accordingly, only a mosaic of normal and dysgenetic gonadal cells would be found in heterozygous carriers of a deletion, even if DSS is essential for ovarian development (see below) and acts cell autonomously.
A double dosage of DSS disrupts testis formation while its absence is compatible with a male phenotype. This paradox could be explained if DSS is a link between the ovarian and testicular pathways. Different genes must be activated and/or repressed to differentiate an ovary or a testis. While it is likely that absence of the SRY gene is sufficient to avoid activation of the testicular pathway in normal females, it is not clear how the reciprocal function - repression of the ovarian pathway - is achieved in normal males. Perhaps DSS is an ovarian differentiation gene with an important function in the sexual determination network. In normal male individuals, ovarian development and DSS function are repressed allowing testis formation. The double dosage of DSS in individuals with Xp duplications and a functional SRY gene, however, hampers repression ofthe ovarian pathway, leading to gonadal dysgenesis and phenotypic sex reversal.
It is puzzling to imagine how, in the presence of X inactivation, the dosage sensitivity of this gene is currently exploited in sex determination; perhaps DSS is a remnant of the ancestral sex determining system which operated by dosage prior to the evolution of X-chromosome inactivation.
Methodology
Clinical details. Patients 711 and BG have been studied previously and found to be duplicated at the ZFX locus4. The extent of the Xp duplications and Yq deletions in patients RR, TMa and SR has also been partially described10. Histology of the gonads and internal genitalia was performed for cases BG, PT and RR. Patient BG has normal Mullerian derivatives, absent left gonad and right streax gonad with primordial sex cords and multifocal gonadoblastoma. Patients PT and RR have both Wofflian and Mullerian remnants and abdominal testes with hypotrophic tubules. Leydig cell hyperplasia was noticed in RR. Patient PT has a slightly hypertrophic clitoris while patient RR has ambiguous external genitalia with perineal hypospadias and a bifid scrotum.
46,XY females were ascertained in Italy. We avoided collecting patients with clinical signs of testicular feminization syndrome (such as presence of well differentiated testes, high testosterone levels, absence of the uterus) but the dinical characterization of some of them was incomplete. Patient B1 has a male karyotype after high resolution banding, is mentally retarded and has minor facial anomalies. She has normal female external genitalia, rudimentary uterus and tubes, remnants of Wofflian structures and streaks gonads.
Molecular methods. Conventional G- and Q- banding and high resolution banding20 was performed for all patients. DNA was obtained from venous blood samples or cultured cell lines, digested with TaqI, EcoRI or PstI, blotted and sequentially hybridized to probes, according to standard procedures2'. The copy number at each locus was determined by the presence of heterozygosity and/or by comparison of band intensity, using the autosomal locus D7S410 (ref. 22) as an intensity control. Band intensiq was measured with the Profil Uno densitometer (Sebia Ciampolini). Construction of the l phage libraries and contigfrom YACs A107E5 and 332F10 was performed as described23.
Acknowledgements
We gregrateful to all investigators who generously provided the probes used in this study and we thank A. Ballabio and P. Goodfellow for thc critical reading of the manuscript. The financial support of Telethon ltaly (Grant n. B.05), Progetto Finalizzato a "Biotecnologie e Biostrumentazione" and "Ingegneria Genetica" of the Consiglio Nazionale delle Riscrche, and of the Commission of the European Communities (Grant GENE-CT93-0027) is gratefully acknowledged.
Received 25 March; accepted 10 May 1994.
REFERENCES
1.