This web page was produced as an assignment for an undergraduate course at Davidson College
Rheumatoid Arthritis
Rheumatoid arthritis (RA) is an autoimmune disease that is caused by chronic inflammation of the joints or in the membrane surrounding the joints (the synovial membrane). This painful systemic disease causes swelling, stiffness, and at times deformity due to irreversible destruction of cartilage, tendons and bones. Figure 1 demonstrates these symptoms and how RA can affect the joints in a person's hands. Figure 2 looks at the hands again in reference to the way RA can cause bone erosion and deformation. Rheumatoid arthritis can also affect other joints in the human body, besides the handsm, over time (see Figure 3). Other symptoms can include a loss of appetite, a low-grade fever, anemia or nodules under the skin, which can add to the cause of deformity. In severe cases this chronic inflammation can also lead to cardiovascular and pulmonary complications and affect other tissues and organs, such as the lungs, eyes, and bone marrow (Ra.com, 2006).
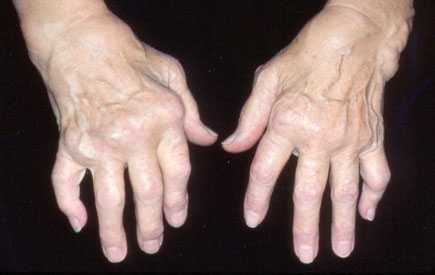
Figure 1.
Advanced stages of rheumatoid arthritis causes swelling, stiffness, and deformity to many joints all over the body like it is shown here in the hands. Courtesy of Cedars-Senai at http://www.csmc.edu/.
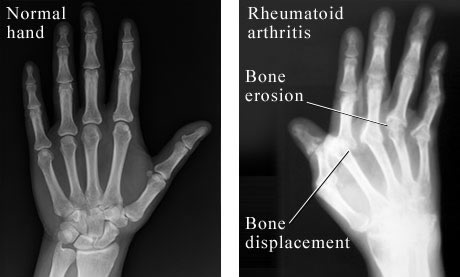
Figure 2. This image shows the contrast of an X-ray of a normal hand on the left to that of a patient with RA on the right. This inflammation, bone errosion, and bone displacement is shown in the right side of the figure. Permission pending from Paul Traughber of Intermountain Medical Imaging at http://www.webmd.com/hw/health_guide_atoz/zm6061.asp.
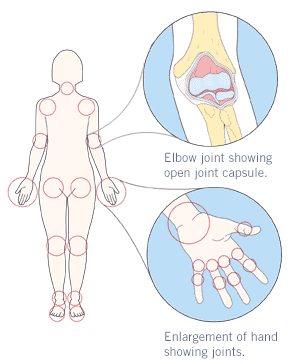
Figure 3. Rheumatoid arthritis affects many joints all over the body, which are are circled in red above. Courtesy of RA.com at http://www.ra.com/ra/rastore/cgi-bin/ProdSubEV_Cat_200635_SubCat_200635_NavRoot_303.htm?#recog.
Rheumatoid arthritis affects approximately 2.1 million people in the United States, which is approximately 1% of the population. This autoimmune disease can affect people of all ages, but typically targets those from 20 to 45 years old. Women are also three-times more likely to get RA than men (Smith, 2006). It has been suspected through various studies that RA is caused by genetic, environmental, and hormonal factors.
Genetic factors
One theory that may cause RA is genetic. A particular study indicates that genetic factors may be of little importance, but it is likely a component of its cause. This study surveyed 37,000 sets of twins and those who had rheumatoid arthritis were placed in clinical examinations. They interestingly found that none of the identical twins both had rheumatoid arthritis and only two of the non-identical pairs both had RA (Svendsen et. al., 2002). According to the study, it appears that RA is not more common in identical twins, compared to non-identical twins. Even though identical twins have the same genes, only one of the twins developed the autoimmune disease. This study cannot disprove a genetic component in susceptibility to RA; however, the results emphasize that the genetic effects are weak compared with environmental ones. Even though RA may not be connected through inheritance, as demonstrated in the twin studies, there is an obvious genetic cause. According to Dr. Howard Smith, 60% of United States’ patients carry the shared epitope of the HLA-DR4 cluster. HLA-DR1 has also been found in a majority of the patients, which adds an extra risk. Other genes within the major histocompatibility complex (MHC) are also involved (2006). Further discussion of these genes and their role in RA are discussed in the section below - Pathogenesis of Rheumatoid Arthritis.
Environmental factors
Some of the environmental factors which can increase the likelihood of having RA have been found by the exposure to bacteria, a particular diet, or smoking. There are numerous infectious agents that may induce RA like Mycoplasma organisms, Epstein-Barr virus, rubella virus, and Streptococcus, among others. In less sanitary environments, people who have been exposed to these bacteria or viruses are more likely to get RA (Smith, 2006). This is also how diets can affect causation of RA. Some researchers have even found that a lack of Vitamin C can increase risk for obtaining RA as well(Warner, 2004). Last, studies show that smoking is a major environmental risk factor. A recent study was conducted involving 930 patients with recent-onset RA and 400 healthy controls from 18 to 70 years of age. From questionnaires about their past smoking habits and a sample of bronchial fluid, researchers found that a history of smoking increases the risk of rheumatoid arthritis (Malattia, 2006).
Hormonal factors
Another theory that affects RA patients is by hormones. Researchers have found that hormones, like estrogen and progesterone, increase during pregnancy and decrease afterward. During pregnancy, women often experience a symptom improvement of RA, but after birth their symptoms will increase again. RA can significantly cause complications during pregnancy through elevated blood pressure, more deliveries by cesarean, and fetal growth retardation in the uterus (Reuters Health, 2006). Because of these hormone differences RA is more prevalent among women, three to one, compared to men.
Pathogenesis of Rheumatoid Arthritis
Autoimmune diseases require that the affected individual have a defect in the ability to distinguish self from foreign molecules. There are markers on many cells that allow the immune system to identify self. However, some markers allow for RA to happen regardless of the markers that may label cells as self. There are obvious factors to rheumatoid arthritis, as listed above, and why it is an autoimmune disease, but the exact cause of this systemic disease is still elusive because several immunopathogenic mechanisms work analogously to cause this autoimmune disease (Smith, 2006). Patients with RA undergo a variation of possible changes and create an initial immune response to a host molecule that uses molecular mimicry to look like a foreign molecule, which the body then attacks. Due to several changes, RA can be categorized as a Type III, immune complex disease, which involves immune complexes containing autoantibodies against soluble autoantigens; or it can be categorized as a Type IV, T-cell mediated disease, which involves an unknown synovial joint antigen as its autoantigen to affect the T cell and antibody-mediated pathways and cause tissue injury (Janeway, 2005). Both factors are important to the pathogenesis of RA.
As an immune complex disease, an autoantibody called rheumatoid factor (shown in Figure 4) has been found among 80% of RA patients (Goldbach-Mansky et. al., 2000). In a study conducted by Goldback-Manskey et. al., a strong correlation between RA and rheumatoid factor was found. There may be other autoantibodies which develop in different stages of the disease, but the rheumatoid factor seems to be the most prominent and prevalent (2000). The rheumatoid factor itself is an “IgM anti-IgG antibody that is produced in normal immune responses” to a severe infection or immunization (Janeway, 2005). T cells are stimulated in an antigen-independent manner, create an abundance of cytokines, interact with enough IgG autoantigen complexes, stimulate ignorant B cells, and then a rheumatoid factor response can be made to bind the Fc fragment of the host's IgG molecule and cause the inflammation that is characteristic of RA (Janeway, 2005).
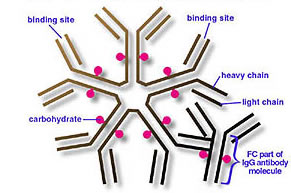
Figure 4. This is the structure of a rheumatoid factor as IgM (rheumatoid factor can also be IgG or IgA). These rhematoid factors are antibodies that bind Fc fragments of the host's IgG molecule and cause an autoimmunity response. This is shown in the bottom right of the figure. Permission pending from Doris Lefkowitz at http://glycoscience.org/glycoscience/document_viewer.wm?FILENAME=G002.
Rheumatoid arthritis is also a T-cell mediated disease, which is the more commonly known consequence for this autoimmune disease. The pathology of RA also extends as a T-cell mediated disease throughout the synovial joint (see Figure 5) along with other organs. In contrast to normal synovial fluid, RA synovial fluid is completely enriched with macrophages, neutrophils, T lymphocytes, and dendridic cells. There are many more cells present in the RA synovial fluid that the joint naturally increases from 1-2 cells in width to 6-8 cells thick. The corresponding inflamed synovial membrane consists of mainly macrophages and T lymphocytes (Feldmann et. al., 1996). Macrophages in particular are the initiators of the pathogenic cascade of RA. Within the synovial tissue, once macrophages are activated they are involved in recruitment and activation of inflammatory cells, cell contact, overexpression of MHC class II molecules, and cytokine production. Macrophages amplify this autoimmune disease by chronic activation of monocytes and production of different cytokines (Kinne et. al., 2000).
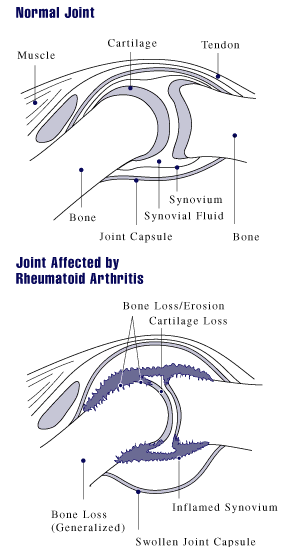
Figure 5. This is view of the difference between a normal joint and a joint affected by rheumatoid arthritis. Pay attention to the synovial fluid and how it changes in the figure from the top to the bottom. The darkened, jagged-edged mass is the inflammation caused by various factors of RA. Courtesy of Wikipedia at http://en.wikipedia.org/wiki/Image:Rheumatoid_arthritis_joint.gif.
On the macrophages, as mentioned above, markers are displayed on cells to recognize self. For RA, these cells are activated and express one of the known clusters of markers is known as the HLA-DR4/DR1 cluster for antigen presentation. In theory, RA seems to require susceptibility to the disease through genetic defects through these markers and an infectious event that triggers an autoimmune response. On one side of the peptide binding groove of HLA-DR, there is a shared epitope made of amino acids 70-74 of the β-chain (Feldmann et. al., 1996). In RA, HLA-DR4 and HLA-DR1 conserve this shared epitope and contribute to the importance of T cell recognition and how this affects the shaping of the T cell receptor (TCR) or presentation of inducing an autoantigenic peptide. Some candidate genes in cytokine polymorphisms are another cause of RA (Feldmann et. al., 1996).
One particular investigation has found a candidate gene with a point mutation in the ζ-associate-protein of 70 kDa (ZAP-70) (Firestein, 2004). This has resulted in causes for abnormal thymic T cell selection and survival of autoreactive clones. In normal immune responses, CD4-associated lymphocyte protein tyrosine kinase (Lck) phosphorylates the ζ-chain of the TCR-CD3 complex. Then, ZAP-70 is recruited to the complex and is phosphorylated by Lck. Then ZAP-70 is used as a linker for activation of T cells, among other signals. However, in a RA patient an amino acid changes from tryptophan to cysteine and leads to spontaneous inflammation that targets joints over a few months, along with rheumatoid factor production. A few other genes may contribute to the abnormal T cell reactivity, but it is unknown at this time. This mutation is a suspected cause for RA, but it could be possible that abnormalities of ZAP-70 are rather caused because of RA (Firestein, 2004).
Another candidate gene of RA, which is widely accepted, has been found in the promoter region of the tumor necrosis factor-α (TNF-α) and Interleukin-1 (IL-1) genes (Kinne et. al., 2000). The proinflammatory cytokines, TNF-α and IL-1, are produced by activated macrophages and fibroblasts in the synovial membrane and are believed to be pivotal cytokines in the signal transduction of the inflammatory cascade. The TNF-α response is thought to come first, and IL-1 expression follows. A mutation in the promoter of TNF-α and IL-1, cause RA patients to suffer from an overproduction of these cytokines and cause articular damage (Feldmann et. al., 1996 and Sebbag et. al., 1997) . From a study by Sebbag et. al., they found that cytokine-stimulated T cells by TNF-α , interacting with macrophages in the rheumatoid synovial membrane, contribute to the continuous and excessive production of TNF-α in the RA joint (1997). It is also suspected that IL-15 may also play a role in inducing macrophage TNF-α production in RA by the activation of T cells (McInnes et. al., 1997). This constant stimulation and signal received to create TNF-α and IL-1 are what induce the chronic inflammation of the joints.
Furthermore, more extensive studies show that the cytokine IL-6 has also been found in the synovial fluid during different phases of RA. IL-6 levels in the synovial fluid correlate to the degree of joint damage of a RA patient and have been thought to promote the generation of osteoclasts. Osteoclases are large multinucleated cells that differentiate from macrophages and break down bone. This reaction may be what promotes excessive bone deformation in the severe cases of the disease in the rise of macrophage activation and fibroblasts in the RA synovial membrane (Kinne et. al., 2000).
Current Treatment for Rheumatoid Arthritis
Many of these possible factors of pathogenesis in rheumatoid arthritis are likely to become therapeutic targets. As autoimmune diseases like RA are studied, more drugs, therapies, and supplements can be administered to treat patients. Many of these treatments have already been created and point towards a successful future. There are no cures for RA, but many of these treatments are minimizing symptoms and stopping the progression of the autoimmune disease. With proper treatment and strategies for joint protection, patients with RA may be able to live a more comfortable life. A summary of the most common types of treatments, which are explained below, are displayed in Figure 6.
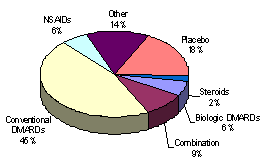
Figure 6. The pie chart above is of different classes of treatment with corresponding percentages. Conventional DMARDs are used 46% of the time, NSAIDs are used 6%, Placebo is 18%, Steroids are 2%, Biological DMARDs are 6%, a combination of treatments is 9%, and other drugs constitutes for 14%. This variety of treatments is better explained below. Permission pending from http://www.metawork.com.
Pain modifiers
The most common administered drugs for RA patients are nonsteroidal anti-inflammatory drugs (NSAIDs). These are the common drugs such as Advil, Motrin, Aspirin, and Aleve. They are used to relieve pain and decrease inflammation (Mayo Clinic, 2006). One of the stronger anti-inflammatory drugs, Vioxx, has however been withdrawn worldwide due to a risk of myocardial infarction and strokes, which is disappointing for RA patients (Singh, 2006). Another common drug used is Celebrex, which is a COX-2 inhibitor and also a NSAID, but less damaging to other parts of the body (Mayo Clinic, 2006).
Some other drugs or supplements beneficial for RA patients are treatments such as taking fish oil, ginger, Valerian, Glucosamine, and Chondroitin. These supplements are herbal medicines that generally improve basic health and subside symptoms. These drugs help to relieve pain, help with sleep, and add necessary vitamins to help in joint strength changes. Likewise, botanical therapies include acupuncture, hydrotherapy, and homeopathy, which all relax the body and can relieve pain (Taibi et. al., 2003). For RA patients it is crucial to live a healthy and relaxing lifestyle by retaining less weight as well because the less weight correlates to less pressure on the joints (Martin, 1998).
Disease modifiers
Another line of therapy, which is maybe one of the most beneficial is called anti-TNF-α therapy (or TNF blockers), which blocks the TNF-α overproduction. This antibody therapy is also considered an anti-inflammatory treatment. Drugs like Humira (adalimumab), Remicade (infliximab), and Enbrel (etanercept) use a monoclonal antibody that inhibits TNF-α so it cannot induce signal transduction for inflammation. Specifically, Remicade has a 90% effectiveness rate in preventing structural damage, reducing symptoms, and reducing progression of joint changes (Shering-Plough, 2006). It does not reduce symptoms or progression 100%, but it is in some ways successful. Likewise, an anti-IL-1 therapy has been created to prevent chronic inflammation. The IL-1 receptor antagonist IL-1Ra blocks signaling through the receptor. In addition, other cytokine antagonists have been used to block the effects of IL-6 in severe cases of RA. This therapy uses a humanized antibody against the IL-6 receptor. This drug is called Kineret (anakinra) (Janeway, 2005 and Mayo Clinic, 2006).
Therapy using an anti-CD4 antibody has also helped the symptoms for a RA patient. This monoclonal antibody causes depletion of helper T cells from peripheral blood (Janeway, 2005). This therapy has not shown promising results, but when combined with anti-TNF-α therapy, treatment has almost blocked the expression of IL-1 completely (Mayo Clinic, 2006). This seems to conclude that combined therapy is a good way to help patients with RA.
Disease-modifying antirheumatic drugs (DMARDs) are also popular for treatment in the early stages of RA. These drugs like minocycline and methotrexate prevent joint and cartilage damage. A few side effects are possible, but with combined therapy the side effects can be eliminated.
Furthermore, one particular study suggests that oral tolerance therapy, consisting of the oral administration of antigens, could alter the immune response given by rheumatoid arthritis (Toussirot, 2002). By taking antigens orally, a state of non or hyporesponsiveness specific for the fed antigen is induced. In this way, the immune system can maintain tolerance to the host’s own antigens in order to avoid immune response against the self determinants. For RA, oral type II collagen autoantigens could be used, but this theory is still new and has only been successful in mice with a 20-30% prevalence of antibodies made. Type II collagen is a likely source for autoantigens because it is the most abundant protein in cartilage. Oral tolerance, like anti-CD4 therapy is used to shift the T helper 1/ T helper 2 balance (Toussirot, 2002).
In extreme cases of rheumatoid arthritis a blood transfusion or bone marrow transplant could be administered to clean out the immune system and aim to rid the body of possible defects that may cause RA. This treatment is not typically given because RA does not usually cause death, but rather shortens life expectancy by 5-10 years (Smith, 2006). In severe cases where life is threatened these treatments can be administered; however, in cases where death is not likely, but deformity has strained the ease of everyday life, joint replacements have been done (Mayo Clinic, 2006).
References
Feldmann, M., F.M. Brennan, R.N. Maini. 1996. Role of cytokines in rhematoid arthritis. Annual Review of Immunology. 14:397-440.
Firestein, Gary S. Aug 2004. The T cell cometh: interplay between adaptive immunity and cytokine networks in rheumatoid arthritis. The Journal of Clinical Investigation. 114(4):471-474.
Goldbach-Mansky, R., J. Lee, A. McCoy, J. Hoxworth, C. Yarboro, J.S. Smolen, G. Steiner, A. Rosen, C. Zhang, H.A. Ménard, Z.J. Zhou, T. Palosuo, W.J. Van Venrooij, R.L. Wilder, J.H. Klippel, H.R. Schumacher, H.S. El-Gabalawy. 9 March 2000. Rheumatoid arthritis associated autoantibodies in patients with synovitis of recent onset. Arthritis Research. 2(3):236-243.
Kinne, R.W., R. Brauer, B. Stuhlmuller, E. Palombo-Kinne, G.R. Burmester. 2000. Macrophages in rheumatoid arthritis. Arthritis Research. 2:189-202.
Malattia, Luca. 2006. The impact of smoking and genes on rheumatoid arthritis. Arthritis & Rheumatism. Accessed 17 April 2006. http://www.xagena.it/news/medicinenews_net_news/64f9f403cbe1b6f341975cfa85484349.html.
Martin, R.H. 1998. The role of nutrition and diet in rheumatoid arthritis. Proceedings of the Nutrition Society. 57(2):231-234.
Mayo Clinic Staff. 3 March 2006. Rheumatoid Arthritis. Mayo Foundation for Medical Education and Research (MFMER). Accessed 22 April 2006. http://www.mayoclinic.com/health/rheumatoid-arthritis/DS00020.
McInnes, I.B., B.P. Leung, R.D. Sturrock, M. Field, F.Y. Liew. Feb 1997. Interleukin-15 mediates T cell-dependent regulation of tumor necrosis factor-alpha production in rheumatoid arthritis. Nature Medicine. 3(2):189.
RA.com. 2006. RA In depth: An up-to-date understanding of Rheumatoid Arthritis. RA.com. Accessed 17 April 2006. http://www.ra.com.
Reuters Health. 3 April 2006. Arthritis, lupus raise pregnancy risks. Arthritis & Rheumatism. Accessed 17 April 2006. http://www.nlm.nih.gov/medlineplus/news/fullstory_31823.html.
Schering-Plough Corporation. 23 March 2006. Remicade approved in Australia for treatment of early rheumatoid arthritis and psoiatic arthritis. Medical News Today. Accessed 22 April 2006. http://www.medicalnewstoday.com/medicalnews.php?newsid=40268.
Sebbag, M. S.L. Parry, F.M. Brennan, M. Feldmann. March 1997. Cytokine stimulation of T lymphocytes regulates their capacity to induce monocytes production of tumor necrosis factor-alpha, but not interleukin-10: possible relevance to pathophysiology of rheumatoid arthritis. European Journal of Immunology. 27(3):624-632.
Singh, Debashis. 9 Oct 2004. Merck withdraws arthritis drug worldwide. British Medical Journal. 329:816.
Smith, Howard R. 6 Jan 2006. Rheumatoid Arthritis. emedicine.com. Accessed 23 April 2006. http://www.emedicine.com/MED/topic2024.htm.
Svendsen, A.J., N.V. Holm, K. Kyvik, P.H. Petersen, P. Junker. 2 Feb 2002.Relative importance of genetic effects in rheumatoid arthritis: historical cohort study of Danish nationwide twin population. British Medical Journal. 324:264.
Taibi, D.M. and C. Bourguignon. 2003. The role of complementary and alternative therapies in managing rheumatoid arthritis. Family and Community Health. 26(1):41-52.
Toussirot, Eric A. 2002. Oral Tolerance in the Treatment of Rheumatoid Arthritis. Current Drug Targest – Inflammation & Allergy. 1:45-52
Warner, Jennifer. 9 June 2004. Vitamin C may fight rheumatoid arthritis. WebMD Medical News Archive. Accessed 24 April 2006. http://www.webmd.com/content/article/88/99940.htm.
Related and Useful Links

Davidson College Immunology Home Page
© Copyright 2005 Department of Biology, Davidson College, Davidson, NC 28036
If you have any questions, comments, or suggestions concerning this page, please contact
Kelly Dresser

{kind=link}