Science . Vol. 263 . 11 FEBRUARY 1994
Click Here to See the Cover Photo
Green Fluorescent Protein as a
Marker for Gene Expression
Martin Chalfie,* Yuan Tu, Ghia Euskirchen, William
W. Ward, Douglas C. Prasher#
A complementary DNA for the Aequorea victoria green fluorescent protein
(GFP) produces a fluorescent product when expressed in prokaryotic (Escherichia
coli) or eukaryotic (Caenorhabditis elegans) cells. Because exogenous
substrates and cofactors are not required for this fluorescence, GFP expression
can be used to monitor gene expression and protein localization in living
organisms.
Light is produced by the bioluminescent jellyfish A. equorea
victoria when calcium binds to the photoprotein aequorin (1). Although
activation of aequorin in vitro or in heterologous cells produces blue light,
the jellyfish produces green light. This light is the result of a second
protein in A. victoria that derives its excitation energy from aequorin
(2), the green fluorescent protein (GFP).
Purified GFP, a protein of 238 amino acids (3), absorbs blue light (maximally
at 395 nm with a minor peak at 470 nm) and emits green light (peak emission
at 509 nm with a shoulder at 540 nm) (2, 4). This fluorescence is very stable,
and virtually no photobleaching is observed (5). Although the intact protein
is needed for fluorescence, the same absorption spectral properties found
in the denatured protein are found in a hexapeptide that starts at amino
acid 64 (6, 7). The GFP chromophore is derived from the primary amino acid
sequence through the cyclization of serine-dehydrotyrosine-glycine within
this hexapeptide (7). The mechanisms that produce the dehydrotyrosine and
cyclize the polyeptide to form the chromophore are unknown. To determine
whether additional factors from A. victoria were needed for the production
of the fluorescent protein, we tested GFP fluorescence in heterologous systems.
Here, we show that GFP expressed in prokaryotic and eukaryotic cells is
capable of producing a strong green fluorescence when excited by blue light.
Because this fluorescence requires no additional gene products from A.
victoria, chromophore formation is not species-specific and occurs either
through the use of ubiquitous cellular components or by autocatalysis.
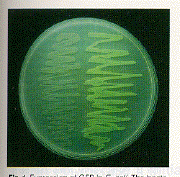
Fig. 1. Expression of GFP in E. coli. The bacteria on the right side of the figure have the GFP expression plasmid. Cells were photographed during irradiation with a hand-held long-wave UV source.
Expression of GFP in Escherichia coli (8) under the control of the T7 promoter results in a readily detected green fluorescence (9) that is not observed in control bacteria. Upon illumination with a longwave ultraviolet (UV) source, fluorescent bacteria were detected on plates that contained the inducer isopropyl-ß-D-thiogalactoside (IPTG) (Fig. 1 ) . Because the cells grew well in the continual presence of the inducer, GFP did not appear to have a toxic effect on the cells. When GFP was partially purified from this strain (10), it was found to have fluorescence excitation and emission spectra indistinguishable from those of the purified native protein (Fig. 2). The spectral properties of the recombinant GFP suggest that the chromophore can form in the absence of other A. victoria products.
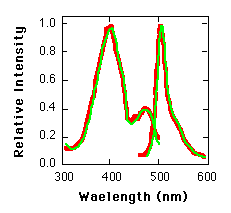
Fig. 2. Excitation and emission spectra of E. coli-generated GFP (red lines) and purified A. victoria L form GFP (green lines).
Transformation of the nematode Caenorhabditis elegans also resulted in the production of fluorescent GFP (11) (Fig. 3 ) . GFP was expressed in a small number of neurons under the control of a promoter for the mec-7 gene. The mec-7 gene encodes a ß-tubulin (12) that is abundant in six touch receptor neurons in C. elegans and less abundant in a few other neurons (13, 14). The pattern of expression of GFP was similar to that detected by mec-7 antibody or from mec-7-lacZ fusions (13-15). The strongest fluorescence was seen in the cell bodies of the four embryonically derived touch receptor neurons (ALML, ALMR, PLML, and PLMR) in younger larvae. The processes from these cells, including their terminal branches, were often visible in larval animals. In some newly hatched animals, the PLM processes were short and ended in what appeared to be prominent growth cones. In older larvae, the cell bodies of the remaining touch cells (AVM and PVM) were also seen; the processes of these cells were more difficult to detect. These postembryonically derived cells arise during the first of the four larval stages (16), but their outgrowth occurs in the following larval stages (17), with the cells becoming functional during the fourth larval stage (18). The fluorescence of GFP in these cells is consistent with these previous results: no fluorescence was detected in these cells in newly hatched or late first-stage larvae, but fluorescence was seen in four of ten late second-stage larvae, all nine early fourth-stage larvae, and seven of eight young adults (19). In addition, moderate to weak fluorescence was seen in a few other neurons (Fig. 3) (20).
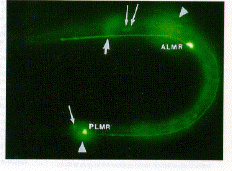
Fig. 3. Expression of GFP in a first-stage C. elegans larva. Two touch receptor neurons (ALMR and PLMR) are labeled at their strongly fluorescing cell bodies. Processes can be seen projecting from both of these cell bodies. Halos produced from the out-of-focus homologs of these cells on the other side of the animal are indicated by arrowheads. The thick arrow points to the nerve ring branch from the ALMR cell (out of focus); thin arrows point to weakly fluorescing cell bodies. The background fluorescence is the result of the animal's autofluorescence.
Like the native protein, GFP expressed in both E. coli and C.
elegans is quite stable (lasting at least 10 min) when illuminated with
450- to 490-nm light. Some photobleaching occurs, however, when the cells
are illuminated with 340- to 390-nm or 395- to 440-nm light (21).
Several methods are available to monitor gene activity and protein distribution
within cells. These include the formation of fusion proteins with coding
sequences for ß-galactosidase, firefly luciferase, and bacterial luciferase
(22). Because such methods require exogenously added substrates or cofactors,
they are of limited use with living tissue. Because the detection of intracellular
GFP requires only irradiation by near UV or blue light, it is not limited
by the availability of substrates. Thus, it should provide an excellent
means for monitoring gene expression and protein localization in living
cells (23, 24). Because it does not appear
to interfere with cell growth and function, GFP should also be a convenient
indicator of transformation and one that could allow cells to be separated
with fluorescence-activated cell sorting. We also envision that GFP can
be used as a vital marker so that cell growth (for example, the elaboration
of neuronal processes) and movement can be followed in situ, especially
in animals that are essentially transparent like C. elegans and zebra
fish. The relatively small size of the protein may facilitate its diffusion
throughout the cytoplasm of extensively branched cells like neurons and
glia. Because the GFP fluorescence persists after treatment with formaldehyde
(9), fixed preparations can also be examined. In addition, absorption of
appropriate laser light by GFP-expressing cells (as has been done for Lucifer
Yellow-containing cells) (25) could result in the selective killing of the
cells.
REFERENCES AND NOTES
1. O. Shimomura, F. H. Johnson, Y. Saiga. J. Cell. Comp. Physiol. 59,
223 (1962).
2. J. G. Morin and J. W. Hastings, J. Cell Physiol. 77, 313 (1971);
H. Morise, O. Shimomura, F. H. Johnson, J. Winant, Biochemistry 13, 2656
(1974).
3. D. C. Prasher, V. K. Eckenrode, W. W. Ward, F. G. Prendergast,
M. J. Cormier, Gene 111, 229 (1992) .
4. W. W. Ward, C. W. Cody, R. C. Hart, M. J. Cormier, Photochem.
Photobiol. 31, 611 (1980).
5. F. G. Prendergast, personal communication.
6. O. Shimomura, FEBS Lett. 104, 220 (1979).
7. C. W. Cody, D. C. Prasher, W. M. Westler, F. G. Prendergast, W.
W. Ward, Biochemistry 32, 1212 (1 993).
8. Plasmid pGFP10.1 contains the Eco RI fragment encoding the GFP
complementary DNA (cDNA) from lgfp10 (3) in pBS(+)
(Stratagene). The fragment was obtained by amplification with the polymerase
chain reaction (PCR) [R. K. Saiki et al., Science 239, 487 (1988)] with
primers flanking the Eco RI sites and subsequent digestion with Eco RI.
DNA was prepared by the Magic Minipreps procedure (Promega) and sequenced
(after an additional ethanol precipitation) on an Applied Biosystems DNA
Sequencer 370A at the DNA sequencing facility at Columbia College of Physicians
and Surgeons. The sequence of the cDNA in pGFP10.1 differs from the published
sequence by a change in codon 80 within the coding sequence from CAG to
CGG, a change that replaces a glutamine residue with arginine. [R. Heim,
S. Emr, and R. Tsien (personal communication) first alerted us to a possible
sequence change in this clone and independently noted the same change.]
This replacement has no detectable effect on the spectral properties of
the protein (Fig. 2). An E. coli expression construct was made with
PCR to generate a fragment with an NheI site at the start of translation
and an EcoRI site 5 ' to the termination signal of the GFP coding sequence
from pGFP10.1. The 5 ' primer was ACAAAGGCTAGCAAAGGAGAAGAAC and the 3 '
primer was the T3 primer (Stratagene) . The Nhe l-Eco RI fragment was ligated
into the similarly cut vector pET3a [A. H. Rosenberg et al., Gene 56, 125
(1987)] by standard methods (26). The resulting coding sequence substitutes
an Ala for the initial GFP Met, which becomes the second amino acid in the
polypeptide. The E. coli strain BL21 (DE3) LysS [F. W. Studier and
B. A. Moffat, J. Mol. Biol. 189, 113 (1986)] was transformed with the resulting
plasmid (TU#58) and grown at 37° C. Control
bacteria were transformed with pET3a. Bacteria were grown on nutrient plates
containing ampicillin (100 µg/ml) and 0.8 mM IPTG. [A similar PCR-generated
fragment (11) was used in our C. elegans construct. As others are
beginning to use pGFP10.1, we have heard that although similar PCR fragments
produce a fluorescent product in other organisms (R. Heim, S. Emr, R. Tsien,
personal communication; S. Wang and T. Hazelrigg, personal communication;
L. Lanini and F. McKeon, personal communication) (23), the EcoRI fragment
does not (R. Heim, S. Emr, R. Tsien, personal communication; A. Coxon, J.
R. Chaillet, T. Bestor, personal communication). These results may indicate
that elements at the 5' end of the sequence or at the start of translation
inhibit expression.]
9. We used a variety of microscopes (Zeiss Axiophot, Nikon Microphot
FXA, and Olympus BH2RFC and BX50) that were equipped for epifluorescence
microscopy. Usually, filter sets for fluorescein isothiocyanate fluorescence
were used (for example, the Zeiss filter set used a BP450490 excitation
filter, 510-nm dichroic, and either a BP515-565 or an LP520 emission filter),
although for some experiments filter sets that excited at lower wavelengths
were used (for example the Zeiss filter set with BP395-440 and LP470 filters
and a 460-nm dichroic or with BP340-390 and LP400 filters with a 395-nm
dichroic). In some instances, a xenon lamp appeared to give a more intense
fluorescence than a mercury lamp when cells were illuminated with light
around 470 nm, although usually the results were comparable. No other attempts
were made to enhance the signal (for example, with low-intensity light cameras),
although such enhancement may be useful in some instances. Previous experiments
had shown that the native protein was fluorescent after glutaraldehyde fixation
(W. W. Ward, unpublished data). S. Wang and T. Hazelrigg (personal communication)
(23) have found that GFP fusion proteins in Drosophila melanogaster
are fluorescent after formaldehyde fixation. We have confirmed that fluorescence
persists after formaldehyde fixation with our C. elegans animals
and with recombinant GFP isolated from E. coli. However, the chemicals
in nail polish, which is often used to seal cover slips, did appear to interfere
with the C. elegans GFP fluorescence.
10. GFP was purified from 250-ml cultures of BL21 (DE3) LysS bacteria
containing TU#58; bacteria were grown in LB
broth (26) containing ampicillin (100 µg/ml) and 0.8 mM IPTG. Induction
was best when IPTG was present continually. Cells were washed in 4 ml of
10 mM tris-HCl (pH 7.4), 100 mM NaCl, 1 mM MgCl2,
and 10 mM dithiothreitol [A. Kumagai and W. G. Dunphy, Cell 64, 903 (1991)]
and then sonicated (two times for 20 s each) in 4 ml of the same buffer
containing 0.1 mM phenylmethylsulfonyl fluoride, pepstatin A (1 µg/ml),
leupeptin (1 µg/ml), and aprotinin (2 µg/ml) and centrifuged
at 5000 rpm for 5 min in the cold. The supernatant was centrifuged a second
time (15,000 rpm for 15 min) and then diluted sevenfold with 10 mM tris
(pH 8.0), 10 mM EDTA, and 0.02% NaN3. Corrected
excitation and emission spectra were obtained with a SPEX F1T11 spectrofluorometer
(Metuchen, NJ) and compared with the purified L isoprotein form of GFP from
A. victoria (M. Cutler, A. Roth, W. W. Ward, unpublished data). The
excitation spectra were measured from 300 to 500 nm with a fixed emission
wavelength of 509 nm, and the emission spectra were measured from 410 to
600 nm with a fixed excitation of 395 nm. All spectra were recorded as signal-reference
data (where the reference is a direct measurement of the lamp intensity
with a separate photomultiplier tube) at room temperature with 1-s integration
times and 1-nm increments. The spectral band widths were adjusted to 0.94
nm for all spectra.
11. Wild-type and mutant animals were grown and genetic strains were
constructed according to S. Brenner [Genetics 77, 71 (1974)1. The plasmid
pGFP10.1 was used as a template for PCR (with the 5 ' primer GAATAAAAGCTAGCAAAGATGAGTAAAG
and the 3 ' T3 primer) to generate a fragment with a 5 ' NheI site (at the
start of translation) and a 3' Eco RI site (3' of the termination codon).
The DNA was cut to produce an NheI - EcoRI fragment that was ligated into
plasmid pPD 16.51 (12, 27), a vector containing the promoter of the C.
elegans mec-7 gene. Wild-type C. elegans were transformed
by coinjecting this DNA (TU#64) and the DNA for plasmid pRF4, which contains
the dominant rol-6 (sul006) mutation, into adult gonads as described
[C. M. Mello, J. M. Kramer, D. Stinchcomb, V. Ambros, EMBO J. 10, 3959 (1991)].
A relatively stable line was isolated (TU1710), and the DNA it carried was
integrated as described by Mitani et al. (15) to produce the integrated
elements uls3 and uls4 (in strains TU1754 and TU1755, respectively).
Living animals were mounted on agar (or agarose) pads as described (16),
often with 10 mM NaN3 as an anesthetic (28)
(another nematode anesthetic, phenoxypropanol, quenched the fluorescence)
and examined with either a Zeiss universal or axiophot microscope. For C.
elegans, a long-pass emission filter works best because the animal's
intestinal autofluorescence (which increases as the animal matures) appears
yellow (with band-pass filters the autofluorescence appears green and obscures
the GFP fluorescence). Because much more intense fluorescence was seen in
uls4 than in uls3 animals (for example, it was often difficult
to see the processes of the ALM and PLM cells in uls3 animals when
the animals were illuminated with a mercuny lamp), the former were used
for the observations reported here. The general pattern of cell body fluorescence
was the same in both strains and in the parental, nonintegrated strain (fluorescence
in this strain was as strong as that in the uis4 animals). The uls4
animals, however, did show an unusual phenotype: both the ALM and PLM touch
cells were often displaced anteriorly. The mature cells usually had processes
in the correct positions, although occasional cells had abnormaily projecting
processes. These cells couid be identified as touch receptor cells because
the fluorescence was dependent on mec-3, a homeobox gene that specifies
touch cell fate (13, 15, 18,28). Expression of mec-7 is reduced in
the ALM touch cells of the head (but not as dramatically in the PLM touch
cells of the tail) in mec-3gene mutants (13, 15). We find a similar change
of GFP expression in a mec-3 mutant background for both uls3 and
uls4. Thus, GFP accurately represents the expression pattern of the
mec-7 gene. It is likely that the reduced staining in uls3
animals and the misplaced cells in uls4 animals are results of either
secondary mutations or the amount or position of the integrated DNA.
12. C . Savage et al., Genes Dev. 3, 870 (1989) .
13. M. Hamelin, I. M. Scott, J. C. Way, J. G. Culotti. EMBO J. 11,
2885 (1992).
14. A. Duggan and M. Chalfie, unpublished data.
15. S. Mitani, H. P. Du, D. H. Hall, M. Driscoll, M. Chalfie, Development
119, 773 (1993).
16. J. E. Suiston and H. R. Horvitz, Dev. Biol. 56, 110 (1977) .
17. W. W. Walthall and M. Chalfie, Science 239, 643 (1988) .
18. M. Chalfie and J. Suiston, Dev Biol. 82, 358 (1981)
19. In adults, the thicker size of the animals and the more intense
autofluorescence of the intestine tend to obscure these cells.
20. These include several cells in the head (including the FLP cells)
and tail of newly hatched animals and the BDU cells, a pair of neurons just
posterior to the pharynx. Expression of mec-7 in these cells has
been seen previously (13, 15). The strongest staining of these non-touch
receptor neurons are a pair of cells in the tail that have anteriorly directed
processes that project along the dorsal muscle line. It is likely that these
are the ALN cells the sister cells to the PLM touch cells [J. G. White E.
Southgate, J. N. Thomson, S. Brenner, Philos. Trans. R. Soc. London Ser.
B 314, 1 (1986)].
21. The photobleaching with 395- to 440-nm light is further accelerated,
to within a second, in the presence of 10 mM NaN3,
which is used as a C. elegans anesthetic (11). However, when cells
in C. elegans have been photobleached, some recovery is seen within
10 min. Further investigation is needed to determine whether this recovery
represents de novo synthesis of GFP. Rapid photobleaching (complete within
a minute) of the green product was also seen when C. elegans was
illuminated with 340- to 390-nm light. Unlike the photobleaching with 395-
to 440-nm light, which abolished fluorescence produced by the 340- to 390-
or 450- to 490-nm light, photobleaching with 340- to 390-nm light did not
appear to affect the fluorescence produced by 395to 490- or 450- to 490-nm
light. Indeed, the fluorescence produced by 450- to 490-nm light appeared
to be more intense after brief photobleaching by 340- to 390-nm light. This
selective photobleaching may indicate the production of more than one fluorescent
product in the animal. These data on GFP fluorescence within E. coli
and C. elegans are in contrast to preliminary studies that suggest
that the isolated native and E. coli proteins are very photostable.
We do not know whether this in vivo sensitivity to photobleaching is a normal
feature of the jellyfish protein (the fluorescence in A. victoria
has not been examined) or results from the absence of a necessary posttranslational
modification unique to A. victoria. or from nonspecific damage within
the cells.
22. Reviewed in T. J. Silhavy and J. R. Beckwith Microbial Rev 49,
398 (1985); S. J. Gould and S. Subramani, Anal. Biochem. 175, 5 (1988):
and G. S. A B Stewart and P. Williams, J. Gen. Microbiot 138, 1289 (1992).
23. R Helm, S. Emr, and R. Tsien (personal communication) have found
that GFP expression in Saccharomyces cerevisiae can make the cells
strongly fluorescent without causing toxicity. S. Wang and T. Hazelrigg
(personal communication) have found that both COOH- terminal and NH2-terminal protein fusions with GFP are fluorescent
in D. melanogaster L. Lanini and F. McKeon (personal communication)
have expressed a GFP protein fusion in mammalian (COS) cells.
24. We have generated several other plasmid constructions that may
be useful to investigators. These include a pBluescript 11 KS (+) derivative
(TU#65) containing a KpnI - EcoRI fragment encoding GFP with an AgeI site
5 ' to the translation start and a Bsm I site at the termination codon.
Also available are gfp versions (TU#60 to TU#63) of the four C. elegans
lacZ expression vectors (pPD16.43, pPD21.28, pPD22.04, and pPD22.11,
respectively) as described (27) except that they lack the Kpn I fragment
containing the SV40 nuclear localization signal.
(NB The Biology Dept. at Davidson College has a form of GFP
that will work in E. coli. If you want to see this for yourself,
let Dr. C. know.)
25. J. P. Miller and A. Selverston, Scence 206, 702 (1 979) .
26. J. Sanbrook, E. F. Fritsch, T. Maniatis, Molecular Cloning: A
Laboratory Manual (Cold Spring Harbor Laboratory Press, Cold Spring Harbor,
NY ed. 2, 1989).
27. A. Fire, S. W. Harrison, D. Dixon, Gene 93, 189 (1990) .
28. J. C. Way and M. Chalfie, Cell 54, 5 (1988).
29. We are indebted to A. Duggan and D Xue for technical suggestions,
to L. Kerr and P. Presley at the Marine Biological Laboratories at Woods
Hole for help with microscopy, to M. Cutler and R. Ludescher for assistance
in obtaining the excitation and emission spectra, to A. Fire for suggestions
on vector construction, and to the colleagues listed in (8) and (23) for
permission to cite their unpublished research. Supported by NIH grant GM31997
and a McKnight Development Award to M.C. and by American Cancer Society
grant NP640 to D.C.P.
15 September 1993; accepted 16 November 1993
M. Chalfie, Y. Tu, G. Euskirchen, Department of Biological
Sciences, Columbia University, New York, NY 10027, USA.
W. W. Ward, Department of Biochemistry and Microbiology, Cook College, Rutgers
University, New Brunswick, NJ 08903, USA.
D. C. Prasher, Biology Department, Woods Hole Oceanographic Institution,
Woods Hole, MA 02543, USA.
*To whom correspondence should be addressed.
# Present address: U.S. Department of Agriculture, Building 1398, Otis Air
National Guard Base, MA 02542, USA.
![]()
![]()
© Copyright
2000 Department of Biology, Davidson College, Davidson, NC 28036
Send comments, questions, and suggestions to: macampbell@davidson.edu