This paper has been reproduced with "permission of Oxford University Press" from an article which appeared in Human Molecular Genetics, 1994, Vol. 3, No. 8, pp. 1437 - 1438.
A novel nonsense mutation in the HMG box of the SRY gene
in a patient with XY sex reversal
Taku Lida1,2,
Yutaka Nakahori1,
Rie Komaki1,
Eai Mori1,
Nobuyoshi
Haysahi3,
Osamu Tsutsumi2,
Yuji Taketani2
and Yasuo Nakagome1,*
*To whom correspondence should be addressed.
1 Department of Human Genetics, School of International Health, University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113,
2 Department of Obstetrics and Gynecology, Faculty of Medicine, University of Tokyo, Tokyo and
3 Department of Gynecology, Okayama Central Hospital, 2-18-19 Honkancho, Okayama City, Okayama 700, Japan
Received May 3, 1994; Revised and Accepted May 20, 1994
The sex determining region Y (SRY) gene initiates sex differentiation in man (1). A small number of cases of XY sex reversal have been described which were due to mutations within the SRY gene, especially in the high mobility group conserved region (HMG box) (2 and 3). We studied the DNA sequence of the SRY gene in 22 Japanese in XY females (manuscript in preparation). Here we report a case who has a single base pair substitution within the HMG box.
The subject was a 28-year-old married Japanese woman with a history of primary amenorrhea and infertility. Physical examination revealed an apparently normal female with a weight of 62 kg and a height of 170.5 cm. While the external genitals were those of a female, they were infantile with no hypertrophy of the clitoris. The vagina was normal and a cervix was present. The uterus was normal in shape and position. Laboratory examination revealed that the amenorrhea was due to ovarian failure. There was no elevation of androgen levels. Repeated chromosome analysis from leukocyte cultures consistently showed a 46,XY karyotype. Both gonads were partially resected since the risk of malignant development in XY gonadal dysgenesis has been reported to be 25 % (4). The gonads consisted of fibroadipose tissue with no malignant cells and nor with ovarian or testicular components. The diagnosis of XY pure gonadal dysgenesis in this case is substantiated by the following features: female phenotype, high stature (+2.9 S.D.), the presence of a uterus and fallopian tubes and an apparently normal 46,XY karyotype.
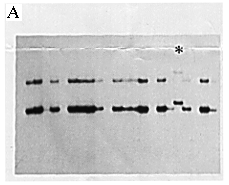
Genomic DNA was prepared from the patient's leukocytes in peripheral blood using the conventional method. The existence of the SRY gene was shown by the polymerase chain reaction (PCR) as described elsewhere (5). We further studied the gene with the single strand conformation polymorphism (SSCP) method (6) and detected an abnormal band pattern (Fig. 1A).
Figure 1A. Detection of a mutation in the SRY gene. PCR-SSCP analysis using a pair of primers spanning the entire HMG box. Each sequences were: SRY 366: 5'-CAGTGTGAAACGGGAGAAAACAGT-3' SRY 367: 5'-CTTCCGACGAGGTCGATACTTATA-3' One of the patients showed an abnormal band pattern (*) in electrophoresis on a 6% acrylamide gel containing 10% glycerol.
Direct sequencing was carried out using, a biotinylated and a non-biotinylated primers which enabled the use of a particular strand of a double-strand DNA molecule for sequencing as only the biotinylated strands bind to the streptavidin coated beads (7). Fig. 1B shows a single base substitution in codon 107 counted from the initiation site of the SRY gene.
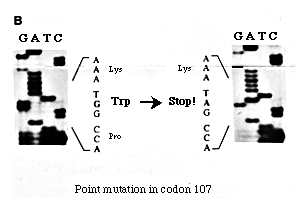
Figure 1. (B) PCR direct sequencing analysis. The left is from a normal male control, and the right is from the patient. The arrow indicates the nucleotide substitution G to A in the 107th codon from the methionine initiation codon of the SRY gene.
Mutations in the SRY gene causing XY sex reversal have been reported (2, 3, 8 and 9). Mutations causing a stop codon within the HMG box have also been reported (10, 11 and 12). The mutation observed in this case also caused a stop codon. However, it is in the middle of the HMG region and different from other mutations reported in the literature (10, 11 and 12).
ACKNOWLEDGEMENTS
Supported by grants from the Faculty of Medicine, University of Tokyo, the Ministry of Education, Science and Culture and the Ministry of Health and Welfare, Japan.
REFERENCES
1. Koopman, P., Gubbay, J., Vivian, H., Goodfellow, P. N., and Lovell-Badge, R. (1991) Nature 351: 117 - 121.
2. Berta, P., Hawkins, J. R., Sinclair, A. H., Taylor, A., Griffiths, B., Goodfellow, P. N., and Fellous, M. (1990) Nature 348: 448-450.
3. Hawkins, J. R., Taylor, A., Goodfellow, P. N., Migeon, C. J., Smith, K. D., and Berkovitz, G. D. (1992) American Journal of Human Genetics 51: 979 - 984.
4. Troche, V., and Hernandez, E. (1988) Obstetrical and Gynecological Survey 41: 74 79.
5. Nakagome, Y., Seki, S., Fukutani, K., Nagafuchi, S., Nakahori, Y., and Tamura, T. (1991) American Journal of Medical Genetics 41: 112 114.
6. Orita, M., Suzuki, Y., Sekiya, T., and Hayashi, K. (1989) Genomics 8: 874 - 879.
7. Hultman, T., Stahl, S., Hornes, E., and Uhlen, M. (1989) Nucleic Acids Research 17: 4937 - 4945.
8. Jäger, R.F., Harley, V.R., Pfeiffer, R. A., Goodfellow, P. N., and Scherer, G. (1992) Human Genetics 90: 350 - 355.
9. Zeng, Y., Ren, Z., Zhang, M., Huang, Y., Zeng, F., and Huang, S. (1993) Journal of Medical Genetics 30: 655 - 657.
10. Hawkins, J. R., Taylor, A., Berta, P., Levilliers, J., Auwera, V., and Goodfellow, P. N. (1992) Human Genetics 88: 471 - 474.
11. Affara, N. A., Chalmers, K. J., and Ferguson-Smith, M. A. (1993) Human Molecular Genetics 2: 785 - 789.
12. McElreavey, K. D., Vilain, E., Boucekkine, C., Vidaud, M., Jaubert, F., Richaud, F., and Fellous, M. (1992) Genomics 13: 838 - 840.
![]()
![]()