This web page was produced as an assignment for an undergraduate
course at Davidson College.
Single-cell RNA-seq
Summary and opinion
Transcription is a dynamic cellular event. In diploid organisms,
the presence of two alleles at heterozygous loci means that
transcription can take place on either homologous chromosome to produce
distinct gene products. Deng et al.
(2014) used single-cell RNA-seq to determine the relative abundance of
transcripts derived from maternal compared to paternal chromosomes in
the early mouse embryo and differentiated tissues. By testing samples
derived from a cross between two genetically defined strains, they
could use SNPs to assign the chromosome of origin for the majority of
transcripts.
Their results are consistent with stochastic gene expression rather
than stable expression of one allele like in imprinting. It is unclear
why they use the term “monoallelic gene expression,” when they admit
that their data is best explained by a non-regulated stochastic
process, which is the null model. I would argue that it is a
misleading—and undoubtedly attention grabbing—use of the term. Here,
for the sake of consistency, I will use “monoallelic gene expression”
to refer to detection of one allele in an experiment, which does not
imply biological regulation. Despite the misleading vocabulary, the
paper does contribute new understanding of the regulation of X
inactivation, which (technical advances aside) is the most interesting
result.
Figure 1
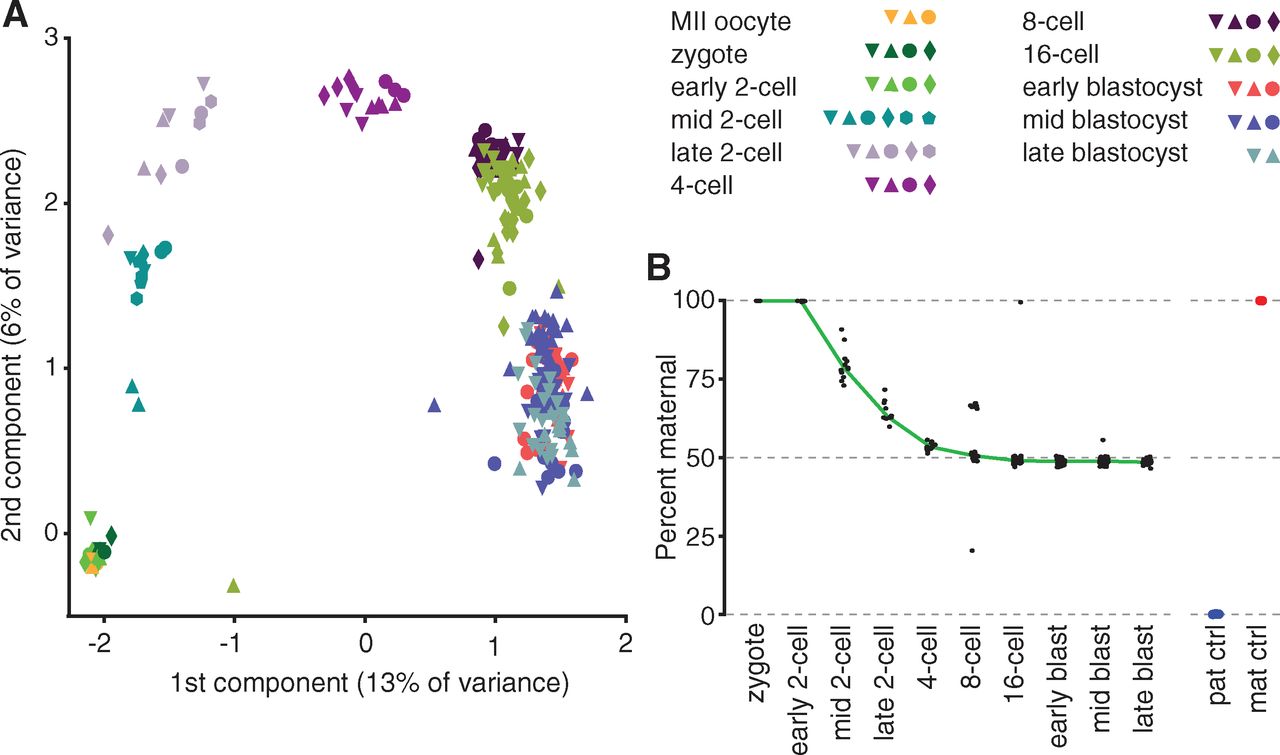
Panel A shows that single-cell transcriptomes cluster
along developmental stages. Each shape is a particular embryo, and each
color is a developmental stage. The single-cell RNA-seq data were
analyzed by principal component analysis, which defines axes (principal
components) that in descending order explain the maximum amount of
variance in transcript levels as possible. Clusters generally contain
cells from several embryos at the same stage, because regulated
patterns of gene expression are fundamental to development.
Panel B shows that by the 4-cell stage, maternal and paternal alleles
are approximately equally represented in the transcriptome. In the
zygote, all RNAs originate from maternal alleles. We can infer that the
paternal pronucleus has not yet fused and become transcriptionally
active. The figure also includes control cells from only the parental
strains, without performing a cross between the two. They found that
their SNP analysis correctly assigns >99% of transcripts to the
correct parent strain of origin when testing the two controls. It was
important that they demonstrate the accuracy of their SNP method before
applying it to determine unknown patterns of gene expression.
Figure 2
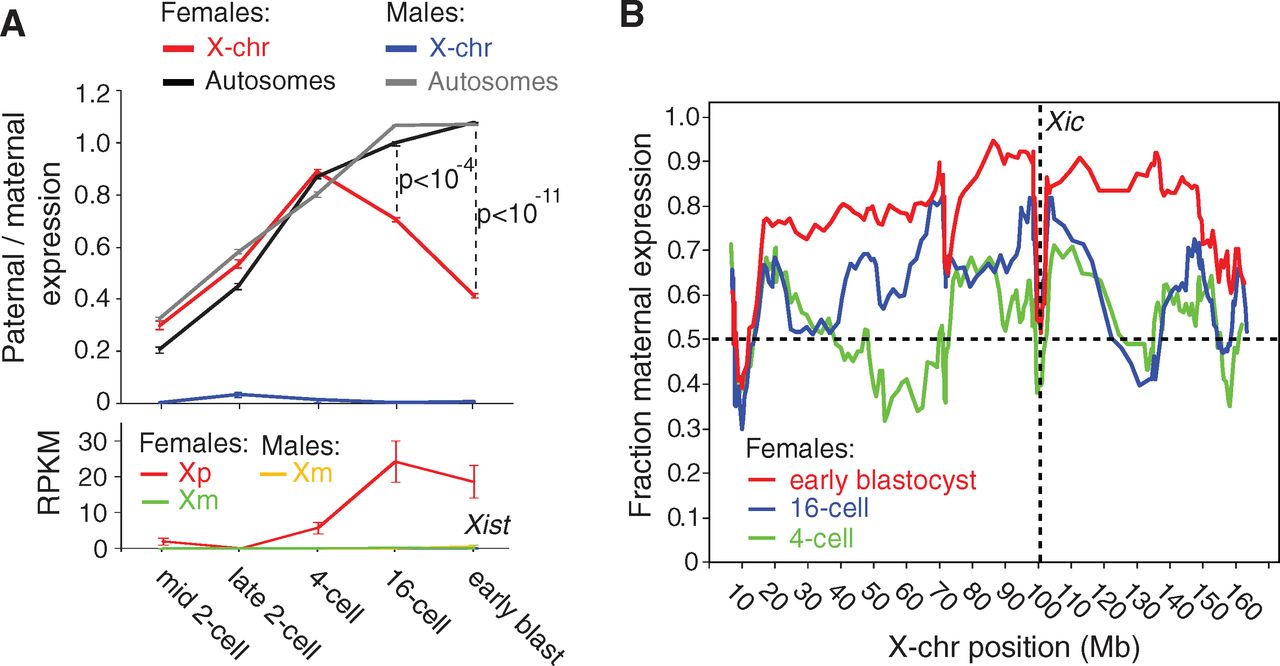
Panel A shows that the paternal X chromosome (Xp)
becomes less transcriptionally active (‘inactivated’) during early
development. A significant bias toward transcription from the maternal
X chromosome appears at the 16-cell stage, and the difference is
greater when development progresses to the early blastocyst. The parent
of origin bias is not present for the autosomes, shown in black and
gray. Consistent with this observed transcriptional bias and the
established role for the Xist transcript in X inactivation, Xist transcription is high during the 16-cell and early blastocyst stages. Additionally, Xist is female-specific and only transcribed from Xp, as the maternal X chromosome Xm will remain active.
Xist is transcribed from the locus Xic, but panel B shows that X inactivation does not simply spread out in either direction from Xic. In fact, loci near Xic
are expressed approximately equally between maternal and paternal
alleles. The observation is shown by the height of roughly 0.5
(fraction maternal expression) for the lines at position 100 Mb, where
the dotted lines in the plot intersect. Although X inactivation does
not spread uniformly from Xic,
it is clear that as development progresses from the 4-cell to 16-cell
to early blastocyst stages, fewer paternal alleles are expressed
overall, shown by red lines above blue and green lines.
Figure 3
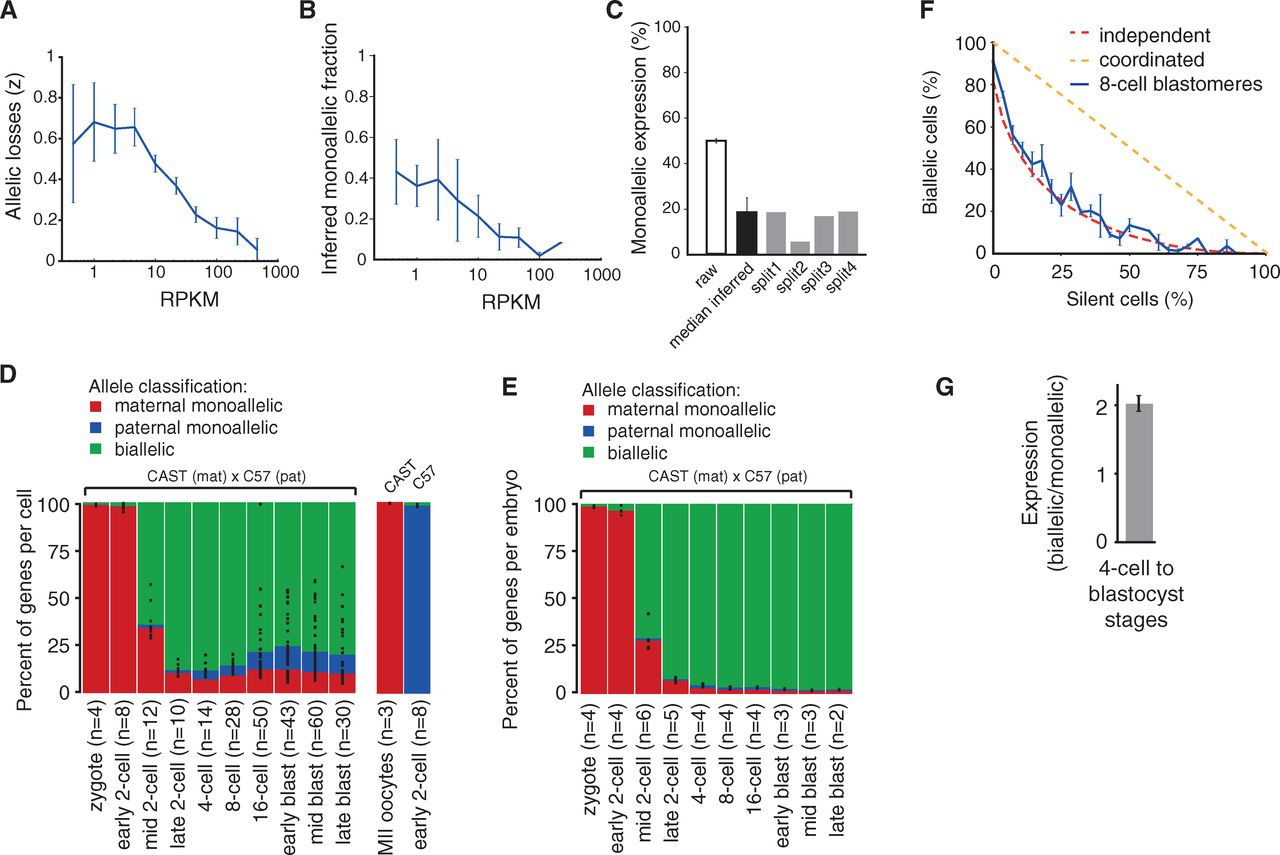
The title of the paper claims an observation of
monoallelic gene expression. However, low levels of transcript could be
lost in the RNA-seq protocol and lead to overestimating the fraction of
genes undergoing monoallelic expression. To determine the efficiency of
their protocol, the authors tested the likelihood of an allele not
being represented in the RNA-seq dataset based on its expression level
(panel A). They split the contents from single cells between two
independent replicates and compared the results. From that, they
inferred that on average 17% of genes showed monoallelic expression,
with more highly expressed genes less likely to show monoallelic
expression (panels B, C).
Panels D and E follow monoallelic gene expression through early
development. Consistent with previous figures, maternal alleles
predominate in early development. However, monoallelic expression of
both maternal and paternal alleles is observed at an apparently high
rate using their single cell method. The fraction of genes undergoing
monoallelic expression varies widely between replicates, particularly
at the 16-cell stage and later.
While it might seem like nearly 25% of genes are expressed from a
single parent during early development, panel E betrays the true
conclusion. In fact, a tiny fraction of genes in the embryo experience
monoallelic expression, besides the strong bias toward maternal alleles
early in development. Panel F shows that their data from the 8-cell
stage closely fit a model of stochastic gene expression. Genes
expressed at low levels, where some cells might not contain transcript
when the analysis is performed, are more likely to show monoallelic
expression. Genes expressed at high levels, where all cells are likely
to contain transcript, are much more likely to have biallelic
expression. In fact, on average, genes with biallelic expression in the
4-cell stage are on average expressed at a 2-fold higher level than
genes undergoing monoallelic expression. The observations are
consistent with stochastic transcription.
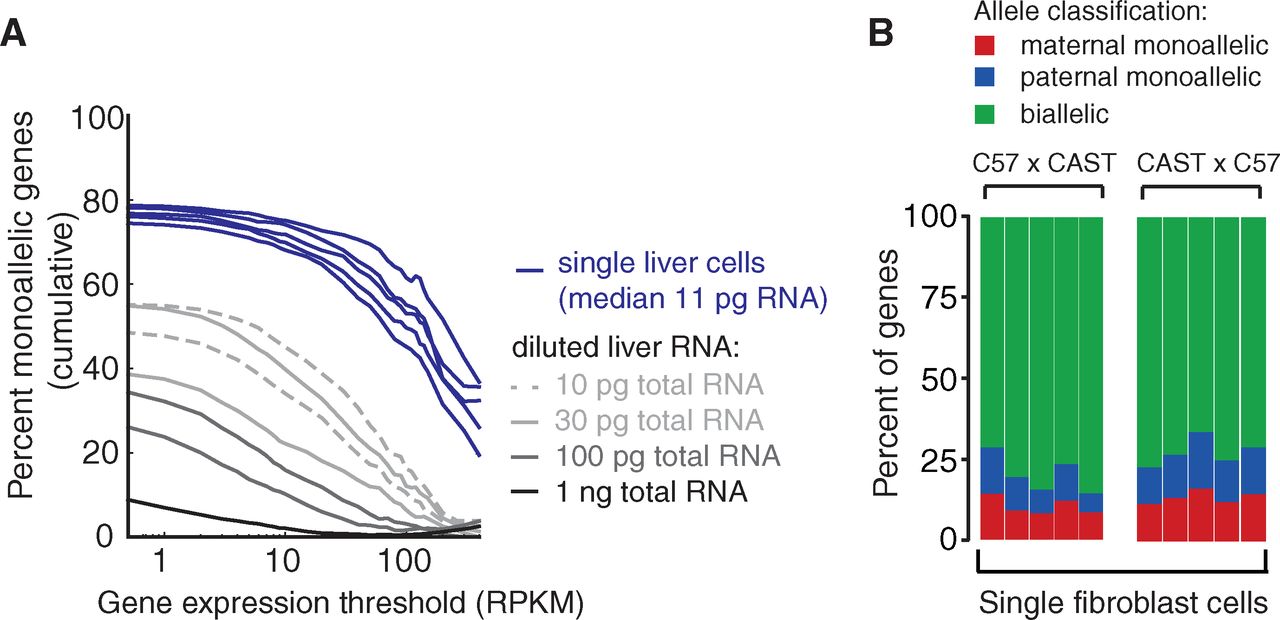
Figure 4
Finally, the authors applied their method to
differentiated liver cells or fibroblasts rather than to early embryos.
Like previously, genes expressed at low levels in the liver are the
majority of genes undergoing monoallelic expression in individual
cells. Simply diluting whole liver RNA extracts replicates the
phenomenon, which is consistent with inherent stochasticity in measure
small levels of transcript. Individual fibroblast cells showed
monoallelic gene expression in proportions comparable to cells from the
early embryo.
Reference:
Deng
Q, Ramsköld D, Reinius B, Sandberg R. 2014. Single-cell RNA-seq reveals
dynamic, random monoallelic gene expression in mammalian cells. Science
343:193-196.
Eric Sawyer's Home Page
Genomics Page
Biology Home Page
Email Questions or Comments.
© Copyright 2014 Department of Biology, Davidson College,
Davidson, NC 28035