This website was produced as an assignment for an undergratuate course at Davidson College
Home safsaProtein dsafOrthologssdaf Jmol Structurefdas Review Paper
What Happens to All That Diet Coke You Drink?
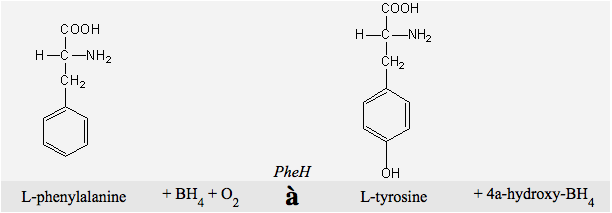
When you drink a diet coke you ingest the amino acid phenylalanine, a component of aspartame, the no-calorie sweetener in the drink. Although phenylalanine is an essential part of your diet, most people get too much and the body must do something to get rid of it all. Phenylalanine Hydroxylase (PheOH) is a tetrameric enzyme which catalyzes the conversion of L-phenylalanine (Phe) to L-tyrosine (Tyr), the reaction is shown below.
Image from Phenylalanine Hydroxylase
Figure 1: The chemical equation PheOH catalyzes.BH4 is a cofactor, and free oxygen is a necessary requirement. The reaction is the simple addition of an -OH group to the 6-ring carbon.
Each of the four subunits of PheOH is comprised of three distinct domains: a 5-kDa C-terminal tetramerization domain, a 33-kDa catalytic domain, and a 12-19 kDa N-terminal regulation domain. The tetramerization domain consists of 2 beta strands which form a ribbon and a long helix. The helical arm of each subunit comes in contact with the other three to form a coiled-coil motif. Coiled-coils form when 2-7 alpha helices wrap around one another forming tightly intertwined portions of the protein. In the center of Figure 2 the four coiled tetramerization domains can be seen, when the coiled-coil forms the subunits are pulled closer together to form the final quaternary structure of the protein.
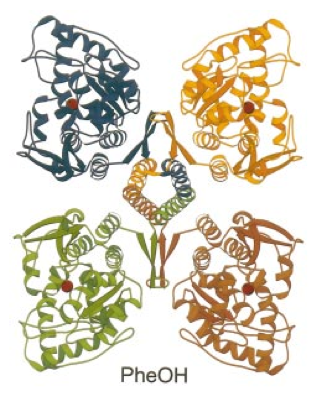
Image from Fusetti et al. 1998.
Figure 2: The four subunits of PheOH. The tetramerization domains can be seen coming together in the very center of the image. The alpha helices form coiled-coils, strong links to hold the enzyme together.
Fusetti et al. (1998) found many biochemical and physical similarities between PheOH and the related enzymes tyrosine hydroxylase (TyrOH) and tryptophan hydroxylase (TrpOH). All three enzymes function to add -OH groups to amino acids. The three proteins share nearly identical tetramerization and regulation domains and differ only slightly in their catalytic domains. “The absence of significant structural differences in the active site of PheOH and TyrOH is not surprising because a similar low substrate binding specificity is observed for the catalytic domains in steady-state kinetic analysis.” (Fusetti et al. 1998) The catalytic domain of PheOH consists of 13 alpha helices and 8 beta strands located deep within the protein. The active site and the area surrounding it is formed by primarily hydrophobic amino acids creating a favorable location for the hydroxylation of the hydrophobic phenylalanine. Each of the subunits contains its own active site allowing the enzyme to catalyze the reaction in four different places. Figure 3 shows one subunit, with the molecule of phenylalanine in the active site. Within the tetramerization domain Fusetti et al. (1998) found two rotation points that allow the protein some flexibility in its form, possibly to allow optimal coiled-coil formation. This flexibility is not found in the similar protein TyrOH. Bergdoll et al. proposed that the rigidity of TyrOH “induces conformational constraints on the peptide backbone that favor the optimal conformation for oligomerization.” (Bergdoll et al. 1997)
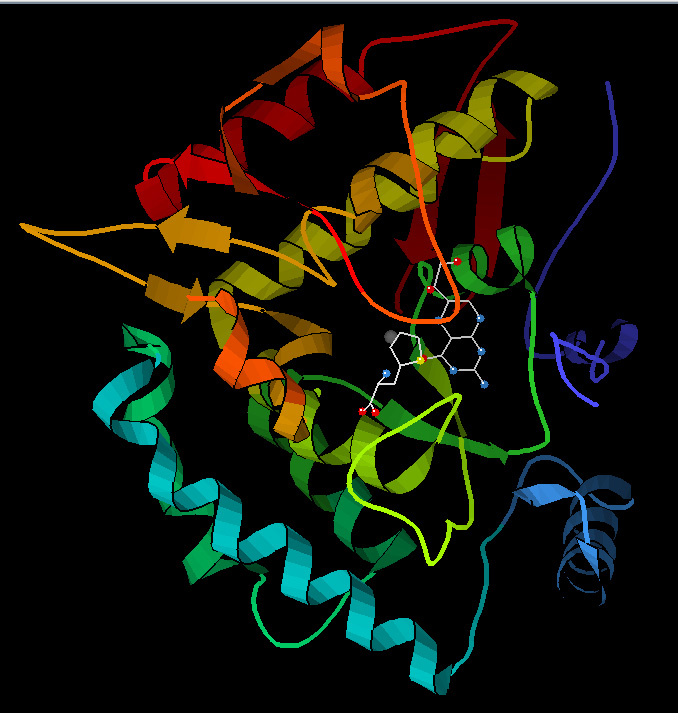
Image from Protein Data Bank
Figure 3: A single subunit of PheOH. The molecule of phenylalanine can be seen in the center, where the catalytic site is located.
How Does PheOH Actually Work?
The exact mechanism of the conversion from phenylalanine to tyrosine is still not known. The conversion of phenylalanine to tyrosine requires the addition of an OH group to the 6-ring carbon. During the reaction the cofactor, tetrahydrobiopterin (BH4) loses two hydrogen atoms to become dihydrobiopterin. BH4 also acts as a reductant, reducing one of the O atoms, while the other atom is added to the carbon ring. In addition to the BH4 an atom of iron is necessary, most likely to somehow stabilize the entire substrate-enzyme complex. Later another enzyme restores dihyrdrobiopterin to tetrahydrobiopterin so the molecule can again act as a cofactor. The rotation sites Fuestti et al. (1998) found may be involved in the cleavage of the H already bound to the carbon ring. The flexibility of that area suggests an ability to change conformation easily. Somehow when phenylalanine enters the active site the H is pulled away from the amino acid, allowing the addition of the -OH group.
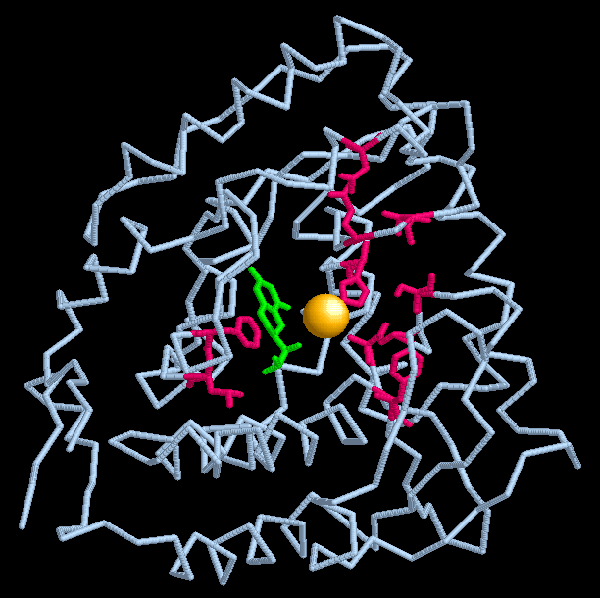
Image From Protein Data Bank
Figure 4: One subunit of PheOH. The iron ion is shown as a gold sphere in the center. The tetrahydrobiopterin cofactor is shown in green. Areas where mutations causing Phenylketonuria frequently occur are shown in pink.
What Determines When PheOH Is Turned On?
The regulatory domain functions to control the activity of the enzyme primarily through physical blockage of the active site with the N-terminus. Phenylalanine, BH4 and phosphorylation all regulate the protein in some way. Phenylalanine acts as the major decider of enzyme function, useful since “it allows the amount of available active enzyme to vary in response to the amount of available substrate.” (Shiman et al.1982) In a normal liver only between 1 and 4% of PheOH is activated. Any growth in phenylalanine concentration increases PheOH activation, up to around 40%. When phenylalanine moves into the allosteric control site “a cooperative physical alteration in the protein occurs which results in the formation of a functional catalytic site.” (Shiman et al. 1990)
Tetrahydrobiopetrin (BH4), like phenylalanine, is both a substrate and a regulator. BH4 can bind to the enzyme to create a PheH‑BH4 complex. When BH4 binds it mostly likely causes the N-terminus region to move further into the active site, effectively preventing phenylalanine from entering. When phenylalanine binds to the allosteric site and induces conformation changes in the enzyme, the bonds with BH4 are broken and the active site becomes available.
The final regulator of PheOH activity is phosphorylation by cAMP dependent protein kinase (PKA). Phosporylation of serine 16 in the regulatory region increases enzymatic activity by causing structural changes which both move the N-terminal region further from the active site and increase the enzyme’s affinity for phenylalanine. Kobe et al. (1999) also postulate that somehow the phosphate aids in phenylalanine’s binding to the active site.
What Happens if Something Is Wrong With the PAH Gene?
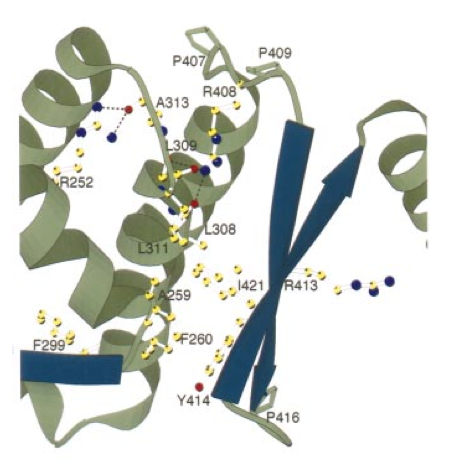
Mutations in the PAH gene cause the metabolic disorder Phenylketonuria (PKU). More information about PKU can be found through the PKU community website. Patients with PKU cannot properly convert phenylalanine so levels of the amino acid gradually increase throughout the body. High levels of phenylalanine in the blood can cause mental retardation when the amino acid accumulates in the brain. Other symptoms of PKU include low levels of pigment in the hair and skin resulting in a light coloration. Currently the United States screens infants for PKU before they leave the hospital so nearly all sufferers begin immediate preventative treatment. Since 1960 more than 150 million babies have been screened and around 10,000 cases of PKU have been detected. PKU currently has no cure and affected individuals must maintain strict diets low in phenylalanine throughout their entire lives. Phenylalanine is a naturally occurring molecule found in common foods such as fish, cheese, milk, meat, and nuts. Due to dietary restrictions PKU patients frequently suffer from various deficiencies. Consequently they must take supplements containing vitamins, minerals, and an amino-acid mixture free of phenylalanine. The majority of mutations that cause PKU can be found in the catalytic domain. A diagram showing some of the most common PKU mutations is below, the mutation sites are shown by white and yellow ball and stick figures.
Image from Fuestti et al. 1998
Figure 5: A closeup of the catalytic domain of PheOH. Common sites of mutation are labeled and shown as white and yellow ball and stick figures.
References
Berdgoll, M et al. 1997 March. Proline-dependent Oligomerization With Arm Exchange. Structure 5: 391-401.
Dutta, Shuchismita and David S. Goodsell. “Phenylalanine Hydroxylase.” Molecule of the Month. January 2005. Protein Data Bank. Accessed 15 February 2010.
Kobe B, et al. 1999 May. Structural Basis of Autoregulation of Phenylalanine Hydroxylase. Nature Structural Biology 5: 442-448.
McDowell, Jennifer. "Phenylalanine Hydroxylase." Protein of the Month. January 2005. European Bioinformatics Institute. Accessed 15 February 2010.
Shiman R, et al. 1982 July. Regulation of Phenylalanine Hydroxylase Activity by Phenylalanine in Vivo, in Vitro, and in Perfused Rat Liver. The Journal of Biological Chemistry 257: 11213-11216.
Shiman R, et al. 1990 June. Mechanism of Phenylalanine Regulation of Phenylalanine Hydroxylase. The Journal of Biological Chemistry 265: 11633-11642.