Affinity Chromatography
PABA standardization
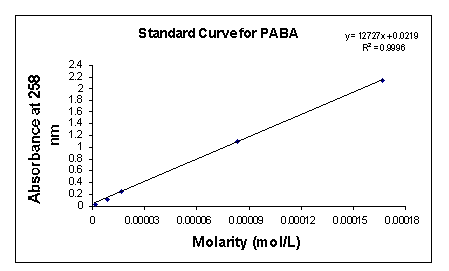
In order to determine unknown concentrations of 4-aminobenzoic acid sodium salt, I generated a PABA optical density (OD) standardization curve (Fig. 3). Serial dilutions of the following molarities: 0.833 mM; 0.167 mM; 83.3 µM; 16.7 µM; 8.33 µM; 1.67 µM; 0.833 µM; and 0.167 µM were made and the OD readings for each of these concentrations taken at 258 nm, the maximum absorption of PABA.
In coupling the PABA sodium salt, I adhered to the BioRad aqueous coupling directions that accompanied the Affi-Gel 10 activated affinity support. I shook the 25 ml slurry of cold (~0° C) activated gel and filtered it using Buchner funnel. I washed the gel cake with multiple volumes of cold deionized water. I transferred the cake into a 125 ml flask along with 25 ml of a 0.15 M PABA sodium salt solution and a magnetic stirrer. The gel slurry gently stirred for 4 hours at 4° C. I filtered the gel again using a Buchner funnel and four volumes of cold deionized water. I scooped the gel into another 125 ml flask and added 30 ml of a 50 mM 3-[N-morpholino] propanesulfonic acid (MOPS) buffer containing 2 percent (w/v) sodium azide. I kept the gel washings for OD analysis. I measured the absorption of the washings and used the standardization plot to find its corresponding molar concentrations. I took all OD readings at 258 nm. After coupling quantification, I gravity packed the gel into a 20 ml affinity column.
Gylcine ethyl ester coupling to an immobilized matrix
I prepared the negative control column using glycine ethyl ester. I adhered to the BioRad blocking procedure for the Affi-Gel 10 activated affinity gel. I shook the 25 ml slurry of the cold gel and filtered using a Buchner funnel. I washed the gel cake with multiple volumes of cold deionized water. I transferred the moist cake to a 125 ml flask and added 2.25 ml of a 1 M glycine ethyl ester solution, 20 ml of cold deionized water, and added a magnetic stirrer to the flask. The gel solution gently stirred for 1 hour at 4o C. After the hour, I added an additional 22.5 ml of 1 M glycine ethyl ester solution. The solution again stirred for 1 hour at 4o C. I filtered the solution using a Buchner funnel and washed with 140 ml of cold deionized water. I transferred the gel cake to another 125 ml flask and added 30 ml of a 50 mM MOPS buffer containing 2 percent (w/v) sodium azide. I gravity packed the gel into a 10 ml affinity column and washed it with deionized water until the gel was free of reactants as detected by OD280 analysis.
Protein Homogenization (Deutscher, 1990)
Prior to protein homogenization, I prepared a 0.5 M dithiothreitol (DTT) stock solution and a 0.2 M phenyl-methyl-sulfonyl fluoride (PMSF) 2-propanol stock solution. The DTT solution was stored at -20o C and the PMSF solution at 4o C.
Figure 4 is a schematic representation of the homogenization procedure. Two rat brains (~ 1.6g each), from Pel Freeze Biologicals (Rodgers, AR), stored at -75o C were quartered and the sections rinsed with 50 ml of chilled buffer A (50 mM Tris-HCl (pH 7.5), 2 mM EDTA, 150 mM NaCl, 0.5 mM DTT). I homogenized the brains on ice with 2.5X (v/m) of chilled buffer B (50 mM Tris-HCl (pH 7.5), 5 mM magnesium acetate, 0.2 mM EDTA, 0.5 mM DTT, 1.0 mM PMSF) in a chilled, glass handheld homogenizer. To homogenize, I used ten strokes for each of the eight rat brain quarters. I decanted the homogenate into two centrifuge tubes and centrifuged them at 15,000g for 30 min using a SW 50.1 rotor in a Beckmann L8-70M Ultracentrifuge. I decanted the supernatant into four ultracentrifuge tubes and centrifuged them at 100,000g for 60 min. A small volume (~400 µl) of the high speed supernatant was removed as a crude homogenate sample. The remaining high speed supernatant portions were divided equally between the experimental and control columns. (Deutscher, 1990).
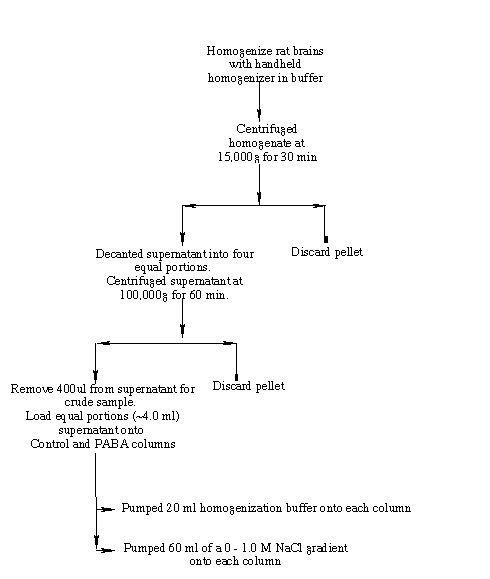
To equilibrate the experimental (PABA) and control (glycine ethyl ester) columns, I washed each with 20 ml of buffer B. Both columns received 4.2 ml of the high-speed supernatant. Following loading of the protein sample, a 20 ml buffer B flow through wash was pumped onto the columns (Fig. 4). A 60 ml 0.0 M-1.0M sodium chloride linear gradient was then pumped onto the columns. 10 ml fractions were collected for the flow through and linear gradient elutions. I carried out the entire procedure at 4o C.
I quantified protein using the Lowry protein determination procedure (Lowry et al., 1951). I set up three sets of seven tubes for standardization ranging from 0 to 100 µg BSA in 5 ug increments. From the standards, I plotted a standard curve of absorbency vs. mg protein. Based on the equation of the best-fit line off the standard curve I calculated the protein concentration of the unknown samples.
Increasing Protein Concentration
I used Eppendorf protein filtration tubes (cat. No. 22 65 040-8) with a 10 kDa filter to reduce the volumes, and thereby increase the concentrations of my protein fractions. I concentrated only those fractions that had concentrations less than 1.0 µg/µl. I centrifuged the samples at 5,000g for 45 to 120 minutes depending on the sample volume I wanted. I centrifuged the samples using a J-20 rotor in a Beckmann J2-21 Centrifuge. I quantified the concentrated protein samples again using the previously described Lowry protocol.
Sodium Dodecyl Sulfate (SDS) Polyacrylimide Gel Electrophoresis (PAGE)
I loaded two 10% acrylamide prefabricated gels (BioRad) with control and experimental quantified protein samples. All samples contained 10 µl SDS loading buffer (47.5% water, 12.5% 0.5 M Tris-HCl pH 6.8, 10% glycerol, 20% SDS (10%) solution, 5% 2-mercaptoethanol, and 5% bromophenol blue (1%) solution), 5 µg protein, and the required amount of SDS running buffer (1.5% (m/v) Tris base, 7.2% (m/v) glycine, and 0.5% (m/v) SDS) to bring volumes to 30 µl. Prior to addition to the gels the respective samples were heated at 95o C for five minutes. Gels were run in SDS running buffer at 200V for 50 minutes.
Coomassie Brilliant Blue R-250 Protein Staining (Deutscher, 1990)
I stained the gels using the Coomassie Brilliant Blue R-250 protein staining method. The 500 ml staining solution contained 0.25% (w/v) CBB R-250, 50% (v/v) methanol, and 10% (v/v) glacial acetic acid. The 1.0 L de-staining solution contained 10% methanol and 5% acetic acid. I incubated the gels in the staining solution for 1 hour before decanting the excess dye and washing with de-staining solution. The gels de-stained for a period of three days after which I took photographs and dried the gels.
Nondiamine Chemical Development Silver Protein Staining (Deutscher, 1990)
After SDS-PAGE, control and experimental gels were soaked in solution 1 (50% (v/v) methanol, 10% acetic acid) for twenty minutes. I then washed the gels for thirty minutes in solution 2 (10% methanol, 5% acetic acid). After washing, I soaked the gels for five minutes in solution 3 (3.4 mM potassium dichromate, 3.2 mM nitric acid) and followed with a rinse of deionized water. I then soaked the gels for 20 minutes in solution 4 (12 mM silver nitrate) after which I incubated the gels in solution 5 (0.28 M sodium carbonate, 0.5 ml formaldehyde per liter). I incubated the gels with three washes of solution 5 making sure to gently agitate during the process. Once the images had sufficiently developed I washed the gels in solution 6 (3% acetic acid). I then briefly washed the gels with solution 7 (10% ethanol) before storing them in solution 8 (7% glycerol, 10% ethanol). Throughout the process I took extreme care in handling the gels and made sure to wear gloves at all times. To determine if excessive background staining originated from protein particular to the prefabricated gels, I poured SDS gels following the Deutscher procedure (1990) and stained them as described above.