Innate immune response
Initial responses in Macrophages and dendritic cells:
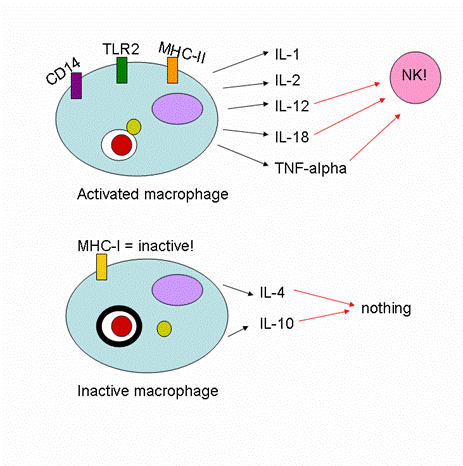
TLR-2 mediates NFkB, and TNF secretion, and binds to the M. leprae and M. tuberculosis wall (2). Some researchers think that the effective functioning of TLR-2 is necessary to the development of the less severe tuberculoid leprosy, in contrast to a systemic lepromatous disease manifestation (2). In lepromatous leprosy, these studies have shown that a significant number of patients have a defective TLR-2 receptor, which then prohibits any cellular immune response from starting (2). Such a lack of cellular immunity apparently causes the M. leprae bacterium to proliferate much more efficiently in the human body (2). TLR-4 may also play a role in M. leprae binding, but to what extent, researchers are not sure (4). The presence of CD14 on the APC surface increases cell mediated responses to M. leprae up to two-fold, but is not necessary for the initiation of the signaling cascade. (2). Studies also show that in lepromatous leprosy, cytokines and chemokines are not released by macrophages - and neither are MHC-I or II significantly expressed (3, 5)! In fact, this inhibition of chemical release may serve instead to enhance bacterial growth (5).
When macrophages (or dendritic cells) do get activated by infection with M. leprae, a number of receptors and inflammatory molecules are upregulated. The interleukins IL-1, IL-2 IL-12, and IL-18 (which acts on both NK and T-cells) are expressed (5). TNF-alpha is secreted in order to activate NK cells (5).
M. leprae induces apoptosis in macrophages in a dose-dependent manner (6). Apparently, this is correlated with high levels of TNF-alpha, as well as various apoptotic receptors (4). It is thought that this apoptosis helps to inhibit bacterial proliferation of the mycobacterium since mycobacteria are not efficient at replicating outside of the cell (4). In fact, researchers claim that this may be one of the main differences between tuberculoid and lepromatous leprosy - this initial clearing of bacterial load through macrophage apoptosis (4).
The cell membrane of M. leprae is highly antigenic, and dentritic cells recognize it readily (6). Upon recognition, the dendritic cells produce IL-12 (6). MMP II is a protein on the surface of M. leprae that stimulates expression of HLA-ABC, HLA-DR, CD86, and CD83 antigens in dendritic cells, and also induces maturation (6). All of this indicates that MMP-II is the protein on M. leprae that binds to TLR-2 on the APC surface (6). Studies also show that the expression of CD1 by dendritic cells is also critical to the activation of T-cells in the later adaptive immune response of the body (7).
The deal is, CD8+ cells are critical to defeating M. leprae once infection has happened (3). The CD8+ cells go around and kill any M. leprae bacteria they can find using perforin and granulysin (3). This is great because they are quite efficient at getting rid of the bacteria that are killing the macrophages (3). However, the T-cells must first be activated – and that is often done by dendritic cells (3). So far, dendritic cells presenting CD1+, CD83+, and the CD40 ligand have been found in areas of M. leprae infection, indicating that they do, in fact, play a significant role in activating T-cell immunity (3).
So far, it is difficult to determine how effective dendritic cells are at activating T-cells specifically during M. leprae infection (3). Studies have shown that infection with M. leprae downregulates the presentation of both MHC-I and II on the surface of dendritic cells, in addition to a lack of significant upregulation of either CD83+ or IFN-gamma (3).
Initial Responses in neutrophils and NK cells:
Both neutrophils and NK cells are very important in inducing further immune response after infection with M. leprae (1). NK cells are required to induce IFN-gamma production, which further activates macrophages and dendritic cells (1). NK cells then help to activate T-cells and promote the differentiation of Th1 cells - a vital part of the defense system against M. leprae infection (1). The activity of NK cells at the site of infection is generally used as a determinant of how successful host defense is, as they serve to both kill infected cells and also heavily activate other members of both the innate and adaptive immune system (1). NK cells also secrete IL-13, though in smaller amounts compared to T-cells (1). This is interesting, because IL-13 is a known cytokine inhibitor, and generally stops any further IFN-gamma production - and therefore essentially stops the inflammatory process (1).
Sources:
1. De la Barerra, S., Finiasz, M., Fink, S., Ilarregui, J., Aleman, M., Olivarez, L., Franco, M. C., Pizzarielao, G., Del Carmen Sasiain, M. 2004. NK cell modulate the cytotoxic activity generated by Mycobacterium leprae-hsp65 in leprosy patients: role of IL-18 and IL-13. Clin Exp Immunol. 135: 105-113.
2. Bochud, P., Hawn, T. R., Adarem, A. 2003. Cutting edge: A Toll-like receptor 2 Polymorphism that is associated with lepromatous leprosy is unable to mediate mycobacterial signaling. J. Imm. 170:3451-3454.
3. Hashimoto, K. Maeta, Y., Kimura, H., Suzuki, K., Masuda, A., Matsuoka, M., Makino, M. 2002. Mycobacterium leprae infection in monocyte-derived dendritic cells and its influence on antigen-presenting function. Infect Imm. 70(9): 5167-5176.
4. Hernandez, M. O., Never, I., Sales, J. S., Carvalho, D. S., Sarno, E. N., and Sampaio, E. P. 2003. Induction of apoptosis in monocytes by Mycobacterium leprae in vitro: a possible role for tumor necrosis factor-a. Imm. 9(1):156.
5. Horwitz, M. A., Levis, W. R., Cohn, Z. A. 1984. Defective production of monocyte-activating cytokines in lepromatous leprosy. J. Exp. Med. 159:666-678.
6. Maeda, Y., Mukai, Y., Spencer, J., Makino, M. 2005. Identification of an Immunomodulating Agent from Mycobacterium leprae. Infect. Immun. 73(7): 4458.
7. Sieling, P. A., Jullien, D., Dahlem, M., Tedder, T. F., Rea, T. H., Modlin, R. L., and Porcelli, S. A. 1999. CD1 expression by dendritic cells in human leprosy lesions: correlation with effective host immunity. J. Imm, 1999, 162:1851-1858.
Copyright Alex Greer 2007
For questions/comments, please contact Immunology professor Dr. Sofia Sarafova at Davidson College. |
