DAB
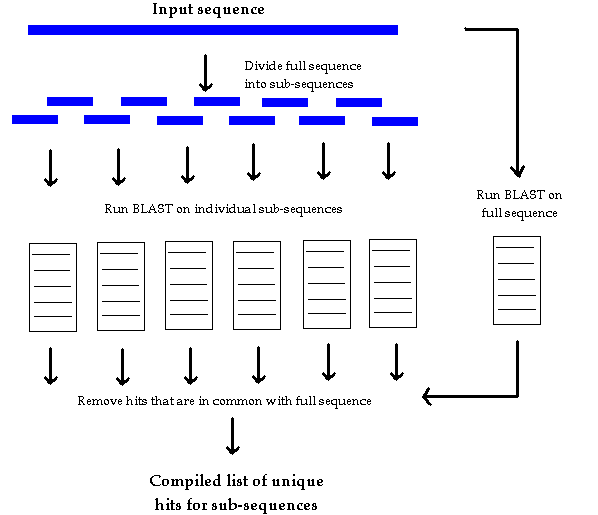
Figure 1. A diagrammatic representation of the Divde-and BLAST
process.
Divide-and-BLAST attempts to address the problem of filtering high similarity hits from a list of hits for a sequence, leaving possibly significant weak similarity hits for further investigation. The program divides its input sequence into a number of sub-sequences, whose length and overlap can be specified as parameters. It then submits both the full sequence and each sub-sequence to the BLAST server using the BLAST network client. After receiving the results, Divide-and-BLAST then removes the hits for the full sequence from the list of hits for each sub-sequence (Fig. 1). The output is a file listing the unique hits for each sub-sequence. If a protein is found to have unique hits on more than one sub-sequence and not show up in the list of hits for the full sequence, it is very likely that there exists a significant similarity between the protein and the input sequence. Even if there are no such duplicate hits between sub-sequences, some of the relatively high similarity unique hits might warrant further investigation using other methods, computational or experimental.
Windows PC:
Unix:
Mac:
Sample files:
A simple input file for testing DAB: idh1_human_short.txt
A sample output for Divide-and-BLAST can be seen here.
These were the results obtained when Divide-and-BLAST was used to analyze the
human isocitrate dehydrogenase protein sequence, using sub-sequences of length
20 amino acids and overlap of 10 amino acids. Notice the hits for isopropylmalate
dehydrogenase; Divide-and-BLAST clearly found an evolutionary relationship,
and localized it to a certain area of the sequence.
What are Expect values?
In general, higher expect values mean lower similarities and vice versa. The
Expect value parameter is the cutoff value -- any hits with Expect values above
the one specified will not be shown. Since Expect value depends on length, sometimes
increasing the Expect value for the sub-sequence BLASTs might turn up more unique
hits than with the default value of 10.0. For a detailed explanation of Expect
values, see the BLAST
FAQ at NCBI.
![]()
![]()
© Copyright 2003 Department of Biology, Davidson College, Davidson, NC
28035
Send comments, questions, and suggestions to: macampbell@davidson.edu