
Background:
In this paper, Stiffler et al. offer a relatively new perspective on protein-protein interaction, also known as interactomics. The authors further characterize the PDZ binding domains found in many signaling and scaffolding proteins. PDZ domains are ubiquitous protein-protein interaction modules 80 to 100 amino acid residues in length that often bind to the carboxyl terminus regions of their target protein.
The abundance of PDZ domains (e.g., the mouse genome contains 928 known domains in 328 proteins) suggests they play an important biological role (Lee & Zheng, 2010). In fact, PDZ domains are known to help mediate cellular processes such as signal transduction and are often localized to multi-protein signaling complexes such as those involved in neuronal synaptic transmission. [Further information about PDZ domains can be found here.]
How are PDZ domains investigated in this paper?
Previously, PDZ domains were thought to fall into one of three functional categories (I, II, & III). However, Stiffler et al. sought to challenge this notion by generating a new model for binding selectivity that could further resolve the PDZ domain-protein interactions. To do this, they first characterized the binding selectivity of 157 PDZ domains in mice using 217 peptides. They used this preliminary data (called their “training set”) to generate a model that could predict additional PDZ domain-peptide interactions. From there, they retested and refined their model in order to further resolve the nature of these interactions, testing its predictability along the way. A more in-depth description and schematic of this process can be found Figure 1A.
Stiffler et al. credit systems biology as providing the paradigm they used to approach their investigation. In general, systems biology seeks to obtain a more holistic understanding of all the components and interactions of a system, rather than the "classical" approach science has primarily embraced that seeks to system by breaking it into individual components--a more reductionist method. Therefore, the researchers sought to understand not just one PDZ domain-peptide interaction, but rather to analyze all the PDZ domains in the entire mouse genome. The goal of such an analysis is to reveal trends that may have been previously obscured by the magnifying glass of reductionism.
Methods:
The researchers first isolated the 157 mouse PDZ domains by cloning, expression, and purification methods. Consequently, they formulated their training set of 217 peptides, acknowledging this compilation may not comprise the full set of ligands that interact with each PDZ domain. The peptides were derivatives of proteins, containing the c-terminus end (due to the PDZ domain preference for this residue) and a fluorescent tag for the purposes of their next step…
Protein microarray
The Stiffler et al. used numerous protein microarrays to assess which of the training set peptides interacted with their 157 mouse PDZ domains. Each array contained four spots of different PDZ domains. Each array was then exposed to a particular peptide to determine with which PDZ domain(s) the peptide interacted. If a peptide bound to a given PDZ domain, the domain's spots would fluoresce after completion of the microarray procedure. [Figure1B provides a representation of their data.]
However, this method is not flawless, of which the researchers were aware. Both false positives and false negatives occur in some frequency with protein microarrays. Therefore, Stiffler et al. implemented another method to address this concern, called quantitative fluoresence polarization:
Quantitative Fluorescence Polarization (FP)
To reduce the number of false negative and positives and thus further refine their model, Stiffler et al. implemented quantitative fluorescence polarization (FP), another method used to analyze protein-protein interactions. In short, FP is based on the premise that fluorescent molecules are generally polarized, and the degree of polarization can be quantified by measuring fluorescent intensity with respect to polarized light. Moreover, a small or unbound molecule will respond differently to polarized light than a larger molecule or complex (such as a protein-protein binding). The image below further describes how FP works:
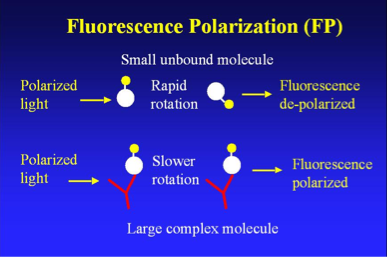
The researchers measured FP at 12 different concentrations of PDZ domains to quantify their dissociation constants (KD), which they used to generate criteria for identifying array false negatives and false positives. This procedure also enabled Stiffler et al. to further refine their model by testing its PDZ domain-peptide predictions with subsequent FP data. [See Figure 1C for FP data.]
Construction & Refinement of the Multidomain Selectivity Model
Using their array and FD data, Stiffler et al. developed a model for predicting PDZ domain-peptide interactions, which they refer to as a multidomain selectivity model (MDSM). The model is based off a previous model (known as PSSM in short), but they note a few key advantages MDSM affords. Unlike PSSM, their new model can resolve the differences among PDZ domains and is based on genomic peptide sequences rather than random ones, thus providing both a more comprehensive and realisitc predictive capacity. However, although MDSM was a modified version of the previously established PSSM, the researchers needed to test the model's ability to perform its intended function (PDZ-peptide binding relationships). This initiated a series of prediction-testing-refinement cycles that led to a refined model they deemed accurate enough to extract meaningful data from regarding PDZ domain-peptide relationships. To do this, Stiffler et al. used the model to predict the false negatives in their array data. Next, they used FP to determine the accuracy of their model, after which they retrained it. This cycle occured three times. The researchers also refined their model to predict which PDZ domains will bind with amino acids at specific positions on the peptides (Figure 2C), one example of the increased specificity of MDSM over previous models. They also clustered different PDZ domain-peptide interactions using a modified version of a cluster algothrithm (Figure 2D), which helped them categorize a range of domain-peptide relationships (again, focusing on the "bigger picture" of interactomics). Lastly, the researchers describe how they further refined their model by implementing a Gaussian kernel smoothing techniqe (to both PDZ domain and peptide data), which served to strengthen the accuracy of their model (Figure 2E).
Implementing MDSM to Predict PDZ Domain-Peptide Selectivity
As we've seen, Stiffler et al. subjected MDSM to many tests and revisions in order to generate a highly refined model that held the potential to share novel insights about PDZ domain-protein interactions. However, the researchers didn't stop there--it was time to test it out. What did they find?
Findings:
In sum, Stiffler et al. extend our understanding of PDZ domain-peptide binding beyond the former, three-class domain categorization. They show that different physiochemical properties of peptides at different positions within their c-terminal residues affect the probability of PDZ domain binding (Figure 3A-D). Furthermore, they model these interactions using three key axes, which, when combined, create a "selectivity space" of interactions, or how different physiochemical and positions influence PDZ domain selectivity. Analysis of their model (Figures 3E-G) provides further evidence that PDZ domains respond to different peptides in a more diverse way than previously assumed that falls along a continum rather than in three distinct classes. However, they also provide evidence that the sequence of the PDZ domain does correlate with its peptide binding preference (Figure 3H). The researchers hypothesize that PDZ domains have evolved increased diversity and specificity in selectivity in order to prevent cross-reactivity, or mis-binding, of proteins and these interaction domains.
Next: Figure Summaries
![]()
Email questions or comments to kaswart@davidson.edu
© Copyright 2011 Department of Biology, Davidson College, Davidson, NC 28035