Focused Reading: See figure 13.8 on page 291.
NOTE: You do not need to wear
gloves while pouring the agar plates, since no mutagenic chemicals
or bacteria are involved in this procedure.
1. Use the work area with a 2.5-3 foot long piece of absorbent
lab counter paper. Label the petri
dishes you will use with your initials and your lab time (e.g.
Tues. Am). I recommend you put your initials on the bottom, along
the perimeter.
2. Look in the water bath on your table for a flask labelled DMA.
This is Davis Minimal Agar
that has been autoclaved to make it sterile, and is being kept
at 47 C to keep it liquified.
3. Think about these important points in pouring a petri plate
before doing it:
a) You must work quickly, because once the container of minimal
agar is removed from the water bath, it will start to harden within
2-3 minutes.
b) When pouring into the petri dish, pour just enough to fill
the dish about half way; this should fill the dish about half
full (or half empty for the pessimists).
c) Although you must work fairly quickly, pour the agar gently
to minimize the number of bubbles (bubbles look amazingly similar
to colonies when the agar hardens).
4. When you are ready to pour:
a) Pull out the container of DMA, remove the cap.
b) Open the cover of the petri dish halfway and pour in the agar
to just cover the bottom of the
dish. Try to minimize the introduction of bubbles.
c) Repeat for all the dishes.
d) Immediately rinse the flask with warm water to facilitate washing
the flask.
5. Let the plate harden 15 minutes before moving it. These plates
will be stored "upside down"
until next week's lab.
Developed from: Dorothy M. Maron and Bruce N. Ames. Revised methods for the Salmonella mutagenicity test. Mutation Research. Vol. 113: 173-215, 1980.
Focused Reading:
"Some chemicals..."pp 277-279 Stop @ "Summary..."
"Point Mutations..." pp 275-277 Stop @ "Chromosomal..."
Web Reading: http://ntp-server.niehs.nih.gov/htdocs/LT-studies/tr389.html
Goals for This Lab:
During this lab, you will pour agar plates to be used
in next week's lab. You will become familiar with the Ames test
which is a world-wide standard for testing new compounds to determine
if they are mutagenic or not. The method you will use this week
was developed here at Davidson College and allows us to screen
more compounds quickly and cheaply. Next week we will use the
traditional method to quatify the degree of mutagenicity for selected
compounds.
NEWS ITEM: April 25, 1997. Dr. Bruce Ames was the Japan Prize ($210,000 cash) for his lifelong work with carcinogens. "When people ask me if I'm the `Ames' of the Ames test, I say: `That was so long ago, that was my father.'" Dr. Ames is professor of biochemistry and molecular biology at UC-Berkeley http://mendel.berkeley.edu/Center.homepage.html and his research is focusing on the relationship of aging, nutrition, and cancer. You can find 4 papers by his group in the April 1, 1997 issue of Proceedings of the National Academy of Sciences in the library.
Our environment is full of potential
carcinogens (cancer-causing agents) such as UV light, industrial
pollutants, pesticides, food additives and tobacco products. These
carcinogens can induce cancers because they are mutagens (chemicals
that cause mutations), which change the nucleic acid sequence
of DNA. It is important to have available a rapid and inexpensive
assay for testing the chemicals we suspect as carcinogenic, as
well as the large number of new synthetic chemicals being produced
each year.
It is estimated that 90% of all carcinogens are also mutagens,
and with this in mind, Bruce Ames and his colleagues developed
a test in the 1970s that uses special bacteria that are very sensitive
to mutagenic agents. The Food and Drug Administration (FDA) now
uses the Ames test to screen many chemicals rapidly and inexpensively.
Those few chemicals that are mutagenic are tested further in animals
to assess their ability to cause cancer.
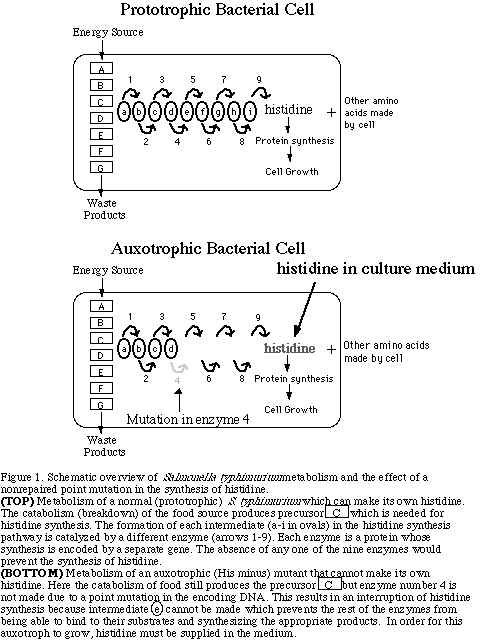
Wild-type cultures of the bacterium Salmonella typhimurium
grow in media without without the addition of any amino acids.
This is possible because they have metabolic pathways for making
all of their own amino acids. Each amino acid has a separate pathway
for its synthesis. For example, Figure 1 shows the pathway for
histidine synthesis, which begins with catabolic intermediate
C and uses nine enzymes (numbered 1-9 in figure 1) to convert
C into histidine.
The Ames test uses a mutant strain of Salmonella typhimurium which cannot grow in the absence of the amino acid histidine because a mutation has occurred in a gene that encodes one of the nine enzymes used in the pathway of histidine synthesis (see Figure 1, bottom). The mutation prevents translation of a functional enzyme #4, and thus the cell cannot complete the conversion of the catabolic intermediate C to histidine. Therefore, the Ames mutants can only grow if histidine is supplied in the growth medium since they cannot make histidine on their own. These auxotrophic mutants are called histidine-dependent or his - (pronounced hiss-minus) mutants because they depend on an external source of histidine to grow. Auxotrophs are mutant individuals that cannot make all the metabolic products that wild-type (prototrophic) individuals of the same species can make.
There are several different mutant strains of S.typhimurium which have different mutations in their DNA. We will use a variety of different mutant strains that have important distinctions that make them suitable for detection of different types of mutagens. Here is a list of the available strains that we can use and what mutation each strain carries:
o TA 1535 has a base substitution that produces a missense mutation in the gene coding for the first enzyme of histidine synthesis. The mutant enzyme has a proline where a leucine is in the wild-type enzyme.
o TA 100 is very similar to 1535, but is also supposed to detect a different range of mutagens.
o TA 1537 has a frameshift mutation (deletion of one nucleotide) in a different gene than is mutated in 1535.
o TA 1538 has a different frameshift mutation (insertion of one nucleotide) in the same gene that is mutated in TA 1537.
o TA 98 is similar to 1538 but is supposed to detect more mutagens than 1538 does.
o TA 102 is significantly different from the others. It has an ochre mutation which means that it has a nonsense mutation. This mutation occurs in the same gene as is mutated in the strain TA 1535.
In addition to the mutations listed above, there are two other shared traits that each of these strains carry. 1) These mutant strains lack a DNA excision-repair mechanism which exists in wild-type bacteria and would normally repair any new mutations in the DNA that are caused by exposure to mutagens during our experiments. The result of this defect is that DNA errors are not corrected, thus enhancing the strain's sensitivity to mutagens. 2) These strains have a defective lipopolysaccharide layer in its cell wall that allows chemicals to penetrate more easily into the cell than is true with wild-type bacteria.
In summary, we have mutant strains of Salmonella typhimurium that cannot synthesize histidine, are very susceptible to additional mutations because they lack the normal repair mechanisms found in bacteria, and are more permeable than wild-type bacteria to external chemicals, including potential mutagens. In order for these cells to survive on a plate that lacks histidine, they must "learn" how to synthesize histidine by undergoing another mutation which corrects the original mutation which prevented the production of the missing enzyme. This is known as a back mutation, or reversion, because this second mutation returns the mutant to the wild-type genotype. This reversion can happen spontaneously due to incorrect DNA replication or as a result of a mutagen, which should more than double the mutation rate over the spontaneous mutation rate. However, note that a spontaneous reversion is NOT the same as a random mutation. The reversion is a specific event that happened randomly but when it happens, you know exactly which bases were affected.
------------------------------------------------------------------------------------------------------------------------------
A brief note about mutations: a mutation is any change in a DNA sequence from the original sequence of nucleic acids, and mutations happen all the time in your cells. Sometimes it is because a mutagen comes from the outside of the cell and in some manner creates changes in the DNA. Often the mutations are just errors that occur during DNA replication when cells divide. In fact, there is an average of nearly one mutation (error) in your DNA every time one cell divides. Your cells have ways to repair the mutated DNA, and they usually do, but if the mistake is overlooked, the change in the DNA is carried on in future replications in the cell. This would be one way that a "spontaneous" mutation can occur, because there was no obvious cause on which to blame the mutation.
------------------------------------------------------------------------------------------------------------------------------
To determine the number of revertants following exposure to a mutagen, we must have a way to differentiate the mutant strain we started with (his- auxotrophs) and the new mutants we may generate (his+ -revertants). The Ames test uses a chemically defined medium for this purpose, meaning the amounts of each ingredient are known, and the medium is lacking one nutrient necessary for bacterial growth. If a his- culture is placed on a chemically defined minimal agar lacking histidine, only those cells that have mutated to his+ (revertants), will grow and form colonies. In theory, the number of colonies that revert and grow is proportional to the mutagenicity of the test chemical.
The chemically defined medium used for the Ames test actually has just a trace (growth limiting) amount of histidine added only to the soft agar overlay. Trace amounts of histidine in the medium are necessary because some mutagenic agents act preferentially on actively replicating DNA. When Ames mutants are plated on this medium, they grow until they run out of histidine (only 2-3 cell divisions lasting about one hour), and the result is a faint, nearly invisible lawn of growth within the overlay. Conversely, revertant bacteria should form large colonies because their growth is not limited since they can produce their own histidine. Each large colony represents one revertant bacterium and its offsping. By definition in the Ames test, a mutagen is any chemical agent which induces twice the number of mutants as occurred spontaneously, and thus is potentially carcinogenic for humans.
What you have read is an overview of the theory behind the Ames test that we will use. Many chemicals in nature, however, are not carcinogenic/mutagenic until after they are consumed by an animal. One job of the liver is to detoxify harmful chemicals but in the process some chemicals are converted into very potent mutagens. For this reason, all test chemicals used in the Ames test by the FDA are routinely incubated with rat liver extract in an oxygenated environment so that liver enzymes, such as oxygenases, will "activate" the chemical being screened; the "activated" chemical is then added to the bacteria. Due to the potential hazards associated with this step, we will not use any liver extracts. Instead, we will test to see if various agents are mutagenic in their "unactivated" states.
NEWS ITEM: Remember the vole found near Chernobyl that was
resistant to mutations and also had high levels of IDH (page 12
of lab manual)? It is speculated that the particular allele carried
by these mutation-resistant voles is more effective at inactivating
oxygen radicals formed by the radiation. Oxygen radicals can "activate"
chemicals and cause them to become mutagenic. Hmmmm.....
The ultimate goal of the laboratory series on the Ames test is to have you design experiments to test an unknown potential mutagen. For the first week, we will use a new variation of the Ames test that was developed at Davidson College during the summer of 1997. This new variation is called the Spot-Overlay Assay and it is designed to allow us to screen a lot of different chemicals quickly and cheaply. Every lab group will use sodium azide as one of its potential mutagens since we know that this is a powerful mutagen (positive control). The second week, we will use the standard overlay assay developed by Dr. Ames to quantitate any mutagens detected by the spot-overlay assay. To successfully perform any Ames test, you will have to be careful to maintain sterile conditions, since you want only Salmonella typhimurium in your petri dish and not other contaminating strains from the air, your fingers, lips ... you get the picture. Furthermore, you must be very careful in this laboratory series:
1) Does "Salmonella" sound familiar to you? It is the bacteria that turns your stomach inside out after eating bad potato salad.
2) You will be handling potential or known carcinogenic/mutagenic materials.
We have faith in your abilities to use your common sense and be careful in what you do with test tubes, pipet tips, etc. that have come in contact with the bacteria and chemicals. Enough warnings, now on to the good stuff.
In your experimental designs for both weeks, the use of controls will be important, just as it is in every experiment. A positive control is a condition that will test positive in your assay; in this case, that means adding a chemical that should allow the cells to grow and reproduce, thus bacterial colonies will grow in a minimal media lacking histidine. Negative controls are conditions that should not cause anything to happen. For our experiments, this would be a condition where no mutations should occur above the rate of spontaneous reversions. It is very possible that more than one positive and negative control might be needed for each chemical and bacterial strain tested.
Perhaps the best way to think about what controls are needed is to look at the possible results for your "test condition" (i.e. what happens when you add your favorite potential mutagen?) and make sure that you could explain the results. For example, what would happen if you saw no colonies in any of your spot-overlays? What if there were bizillions of cells in every spot-overlay? Your control condition spot-overlays should be designed so that you can interpret these results.
How will you determine the "spontaneous" mutation rate? Once you decide this, you can look at the possible outcomes of this experiment. There are only two: either you will see more colonies with a potential mutagen present, or you won't. Take the first possible outcome: if you see more colonies in your spot-overlay with a potential mutagen (due to new mutations) than you see in the spot-overlay with no added chemicals, can you be sure that it was due to the potential mutagen and nothing else? What control(s) do you have to perform to make sure this is true?
Now the second scenario: let's say you get few if any colonies with your potential mutagen present. Can you be sure that this chemical is not a mutagen? What other variables could be different, or go wrong, to give the result of no colonies? If you were testing bleach and no colonies grew, what could you conclude?
After thinking through the possibilities and discussing them in class, we will have several controls, both positive and negative, to include in our experimental design for this first week.
o CAUTION!! (Do I have your attention?) Pay attention to what you are handling, and if it is a container with the bacteria or the chemicals, be sure and wear gloves. PLEASE have only those people handling these things wear gloves, to conserve their use, and you can use 1 pair for the entire lab period.
1) Be aware of where you throw away used gloves, pipet tips, etc. that have come in contact with bacteria and chemicals BEFORE you use them. All trash, including tubes with bacteria and pipet tips go in the orange biohazard bags, which will be autoclaved. The metal caps are cleaned and reused.
2) When handling anything that is supposed to be sterile, such as the pipet tips, petri plates, tubes of distilled water, bacteria, agar, etc., be sure to uncover or uncap itmes for as brief a time as possible. Also, be sure and keep the caps of tubes and lids of petri plate facing down towards the floor when you are holding them to reduce the possibility of contamination.
1. Obtain six pre-poured petri plates with minimal agar (these
have no histidine at all). Label on the bottom plate's edge (where
arrow is in diagram below) your group name, the date, and what
strain of cells will be tested. Divide each plate into 6 equal
parts. Label each pie-shaped section with the chemical to be tested
in that area. Labeling petri dishes is tricky: I recommend writing
small, and around the perimeter of the bottom.
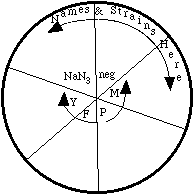
2. Locate the tubes that contain the soft-agar overlay in the water bath on your table - each group will have 36 (6 plates with 6 spots on each) of these. These tubes have been autoclaved to sterilize them and put at 47° C to keep them liquified.
3. Locate the tubes containing the different strains of Salmonella which will be kept at room temperature on the lab bench.
4. If you will be touching tubes, pipets, or plates that contain bacteria, you must PUT ON GLOVES.
5. Locate your chemical to be assayed and any solvents you may be adding to the overlay soft agar tubes.
NOTE: Now you must work quickly to make sure the soft agar overlay does not harden in the tube. Make sure you understand all aspects of step 6 before proceeding.
6. Sterilely add your test chemical and bacteria to the overlay tube and spot this onto the minimal agar.
a) Set your pipettor to the appropriate volume of test chemical (to be determined after discussions with your instructor). Open the box of sterile pipet tips, aseptically put one on, and take an aliquot of your test chemical (or solvent used as a control) into the micropipettor.
b) Remove the cap from the overlay tube, and discharge your aliquot of the test chemical into the overlay tube.
c) Using one strain at a time, swirl the tube containing the bacteria, remove 60 µl of S. typhimurium tester strain and aseptically add it to the overlay tube from step b above.
d) After swirling the tube to mix the bacteria and test chemical, withdraw 200 µl of the mixture.
e) Lift the top of the petri plate (open end facing down) and quickly but gently discharge the bacteria/chemical/agar onto the minimal agar - you should create a small puddle.
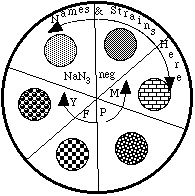
f) Repeat this step until each strain has been tested with each potential mutagen.
g) Invert the plates (i.e., store with the lid on the bottom) and place in a 37° C incubator for 48-72 hours.
After the appropriate incubation period, the plates will be removed from the incubator and store at 4° C until next week. At that time, you will count the number of visible colonies in each spot-overlay, including your negative control spot-overlay. Even though the size of the colonies may vary greatly, they still arose from only one bacterium that has reverted, thus "all colonies are created equal". A colony consists of a distinct white spot that you can clearly distinguish from a bubble or other similar looking phenomena. Your definition of a colony (anything that shows up vs. only really big ones, etc.) is less important than being consistent in counting colonies from one plate to another. It should only take you 15 to 30 minutes to count. When you have finished counting, put the plates in the refrigerator since refrigeration will slow their growth.
NOTE: Be prepared to list the number of colonies in your test conditions and controls for the class to analyze next week. We may look at the plates during the next lab period so please do not throw them away yet.
A. Equipment
Incubator at 37° C
Water bath at 47° C
Micropipets of 1-20 µl, 20-200 µl, and 200-1000 µl
Sterile pipet tips
B. Materials and Reagents
o Petri plates of Davis Minimal Agar (DMA), about 25 ml/plate
o Tubes of top agar (0.6% DMA, 0.5% NaCl, 0.05 mM biotin, 0.05
mM histidine)
o 16-24 hour culture of S.typhimurium, strains TA1535,
TA1537, TA1538, TA97a, TA98,
TA100, TA102, grown in histidine containing medium.
o sodium azide (NaN3) at 0.1 µg/ µl
o Any potential mutagens that you had prepared in advance (must
be sterilized).
o Sterile dH2O or other solvents
1. Ames, B., McCann, J. and Yamasaki, E. Methods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian Microsome Mutagenicity Test. Mutation Research 31: 347-364, 1975.
2. Ames, B, and Yanofsky, C. The Detection of Chemical Mutagens with Enteric Bacteria, pp. 267-282. In: Chemical Mutagens-Principles and Methods for their Detection, Vol. 1, A. Hollander (Ed.), Plenum Press, N.Y., 1971.
3. Maron, D. R. and Ames, B. N. Revised methods for the Salmonella mutagenicity test. Mutation Research. Vol. 113: 173-215, 1980.
1) For next week, we will quantify the mutagenicity of any test chemical that your preliminary results suggest are worth further study. You will design the experiments at that time and perform the more traditional method of plate incorporation for the Ames test.
© Copyright 2000 Department of Biology,
Davidson College, Davidson, NC 28036
Send comments, questions, and suggestions to: macampbell@davidson.edu