The field of biotechnology is growing quickly; its tools are
powerful and its impact is increasingly evident in news, medicine,
and daily life. Biology teachers would like to share the process
and the methods of biotech research with students, but the complicated
nature of the procedures can make designing laboratory courses
difficult. We have developed a biotechnology laboratory course
for undergraduate students that can make it much easier to teach
biotech methods and scientific thinking. This course teaches current
techniques as students progress through a cloning and expression
project. The core
schedule requires eleven weekly three-hour sessions, but provides
many choices of preparation and analytical procedures. A range
of optional activities may be added, depending on the resources,
interests, and focus of the class. Students can research the project
through genomic databases and articles before beginning the project
and/or they can carry out several types of molecular analysis.
Development of this laboratory course is part of an ongoing process
to design a set of biotechnology-related laboratory courses that
focus on one enzyme, isocitrate dehydrogenase (IDH). Cell, molecular,
and developmental biology, biochemistry and genetics laboratory
courses can each analyze IDH using theory and methods unique to
that area of biology. We hope that students will realize that
there are many ways to analyze a single gene product and that
it is important to retain knowledge from one laboratory experience
to another. We selected IDH because it is well-characterized,
ubiquitous, important to cellular function, and familiar to students.
It is an excellent protein to study in the laboratory because
it is non-toxic, easy to purify, and has several testable variables.
IDH catalyzes the oxidative decarboxylation of isocitrate to 2-oxoglutarate,
providing both NADPH and 2-oxoglutarate for necessary metabolic
and amino acid synthesis activity. The particular IDH isoform
cloned in the project outlined below is yeast (Saccharomyces
cerevisiae) cytosolic NADP+ -dependent isocitrate dehydrogenase
(IDP2). Yeast cells contain three genetically independent isoforms
of IDH: IDP2, and two mitochondrial forms, NAD+- and NADP+-dependent
isocitrate dehydrogenases (IDH and IDP1, respectively) (Loftus
et al., 1994).
This laboratory course allows students to be involved in all facets
of the research process. The project's main purpose is to clone
and express yeast cytosolic NADP+ -dependent isocitrate dehydrogenase
(IDP2). Unlike most higher organisms, yeast genes do not contain
introns, allowing cDNA to be cloned for expression directly from
genomic DNA. Major steps in the process include using polymerase
chain reaction (PCR) to amplify the IDP2 gene, IDP2, from
yeast genomic DNA, ligating it into a cloning vector, transferring
the gene to an expression vector, then expressing and analyzing
the recombinant protein (Figure I). When the expression vector
transcribes the IDP2 mRNA, a twelve codon chain is added
to the 5' end, called a T7 tag. Translation of this mRNA creates
an IDP2 protein with a T7 epitope-tag on the N-terminus, easing
detection of the protein on an immunoblot.
We will provide interested users with course specific recombinant
plasmids (pCR2.1-IDP2 and pET5a-IDP2F and -IDP2R)
as insurance. By providing pre-made constructs of recombinant
vectors, we hope to create a safety net for teachers and students.
Students' small but critical mistakes will not prevent the class
from finishing the semester's "project," as there will
always be a back-up construct with which to proceed. All materials
which we do not provide can be purchased from major biotechnology
or chemical companies.
Strains, Vectors and Reagents
The protease deficient yeast strain JYW2878 was obtained from Dr. John L. Woolford at Carnegie Mellon. Reagents and protocols for polymerase chain reaction (PCR) and ligation of the pCR2.1 vector are components of the TA Cloningª kit obtained from Invitrogen. Competent cloning strains of E. coli, Top10F' and JM109 were also from Invitrogen. The pET5 expression vector system, including BL21(DE3) expression cells, materials and protocols for transformation, expression and collection of protein were obtained from Novagen. All restriction enzymes and calf intestinal alkaline phosphatase were obtained from Promega. All other reagents are available from Sigma Chemical Company and/or Fisher Scientific unless otherwise noted.
Primer Design
The yeast cytosolic NADP(+)-dependent isocitrate dehydrogenase gene (IDP2) was previously isolated and cloned (Loftus et al., 1994). We used the gene sequence published at that time to create primers which would amplify the 1.2 kb IDP2 gene sequence using PCR. Primers were engineered to amplify the open reading frame of the gene, placing a BamHI site directly before the start codon, and a HindIII site directly after the stop codon (Forward: 5' G C A T G G A T C C A T G A C A A A G A T T A A G G T A G C 3' ; Reverse: 5' T C C G A A G C T T T T A C A A T G C A G C T G C C T C G A 3' ).
Cloning into pCR2.1, the TA Cloningª vector
The only deviations from Invitrogen's PCR protocol which we found useful were the inclusion of 3% DMSO and the addition of Taq polymerase after the reaction mixture had been heated to 95° Celsius for 4 minutes (hot start). Product from the primary PCR was then used as template for a secondary PCR to further amplify the proportion of IDP2 in the product. This secondary PCR product was purified using a phenol/chloroform and ethanol extraction (Sambrook, et al., 1989). The PCR product was then ligated directly into the pCR2.1 plasmid using the adenine overhangs added to the 3' ends of the PCR product by Taq polymerase. Top10F' E. coli competent cells were transformed with the pCR2.1-IDP2 ligation product, and plated on LB ampicillin (100 mg/ml) plates containing IPTG and X-gal (Sambrook, et al., 1989). Twelve white colonies were chosen for screening and grown overnight in 2 ml LB-ampicillin cultures. Plasmid DNA was isolated from these cultures using an alkaline lysis procedure (Sambrook, et al., 1989). The isolated plasmid DNA was analyzed by restriction digestion with BamHI and HindIII followed by agarose gel visualization to ascertain if the colony contained the 1.2 kb IDP2 gene sequence in the pCR2.1 vector. Colonies containing the recombinant plasmid were grown in 100 ml liquid cultures of LB-ampicillin overnight and the plasmid DNA was isolated using a Qiagen plasmid purification kit and stored at -20° Celsius. Plasmid DNA concentration was determined using a OD260 measurement on a Perkin Elmer Lambda 3B UV/VIS spectrophotometer.
Cloning into pET5a, the expression vector
The pCR2.1-IDP2 DNA was prepared for ligation into the expression vector pET5a by digestion with BamHI, separation from pCR2.1 by agarose gel electrophoresis and recovery by electroelution in a "V" apparatus (Medical Specialties). Briefly, the electroelution process uses current to draw the target DNA out of a small cube of agarose gel and into a vertical V-shaped well containing 7.5 M ammonium acetate. DNA is precipitated from the viscous ammonium acetate solution using 10% 0.3 M sodium acetate and glycogen in an ethanol precipitation procedure (Sambrook, et al., 1989). The pET5a vector was also digested with BamHI and treated with calf intestinal alkaline phosphatase (CIAP, Promega) to prevent self-ligation. The digested IDP2 fragment was ligated into the CIAP-treated pET5a plasmid and pET5a-IDP2 ligation product was used to transform JM109 E. coli competent cells by heat-shock. Transformed cells were plated on LB-ampicillin plates, and all colonies that appeared were screened as described above, using an EcoRI digestion. Digestion with EcoRI was designed to reveal both whether a colony contained a pET5a-IDP2 recombinant plasmid and in which orientation the IDP2 gene was ligated. The plasmid DNA of one "forward" clone and one "reverse" clone was harvested using a small scale alkaline lysis procedure (Sambrook, et al., 1989). This purified plasmid DNA was used directly to transform an expression strain of E.coli, BL21(DE3). These transformants were also plated on LB-ampicillin agar plates and one colony from each transformation was screened to confirm the presence of the correct plasmid using an EcoRI digestion.
Expression and Isolation of IDP2 protein
In the expression phase of the experiment, the clone containing IDP2 in reverse orientation was used as a negative control as it expresses pET5a's T7 tag (Figure 5) but contains a stop codon soon thereafter, resulting in a 36 amino acid peptide. Three 2 ml liquid cultures of one forward clone and three 2 ml cultures of the reverse clone in exponential growth (OD595=0.5) were induced to express the contents of the plasmid in 0.4mM IPTG for one hour. Cells were centrifuged after the induction period for 5 minutes in a microcentrifuge (13,000 rpm), and the medium of each was aspirated. Each pellet of cells was then resuspended in 0.2 ml cold 50 mM Tris HCl pH 8.0 and 2 mM EDTA. Lysozyme was added to a concentration of 100 mg/ml and Triton-X 100 to 0.1%. The cells were incubated in the lysing reagents for 15 minutes at 30° C. After a brief cooling period on ice, the lysates were treated to eliminate DNA contamination in pairs of forward and reverse products. DNase was added to 100 mg/ml to one set and incubated at 37° C for 45 minutes. A second set was treated with 10 mg/ml DNase and incubated at 37° C overnight (19 hours). A third set was mechanically sheared using a 27.5 gauge needle, through which the entire volume of lysate was drawn up into a 3 ml syringe and expelled 50 times. After the DNA degradation treatments, the lysate protein was precipitated by centrifugation for 15 minutes in a microcentrifuge (13,000 rpm). The protein pellets were resuspended in 10 ml distilled water and stored at 4° C.
Protein Analysis by Immunoblot
Polyacrylamide gels were cast and run using Mini PROTEAN II equipment (BioRad). A 7.5% SDS-polyacrylamide resolving gel was cast under a 4% stacking gel (Deutscher, 1990). Proteins were prepared by addition of SDS sample buffer (0.06 M Tris HCl, pH6.8, 10% glycerol, 0.025% bromophenol blue, 1% SDS and 5% B-mercaptoethanol). The samples were loaded onto the gel, run at 100V for approximately 20 minutes through the stacking gel, and at 200V for approximately 35 minutes through the resolving gel. Prestained low molecular weight markers were used (BioRad). The gel was then equilibrated for 15 minutes in Western transfer buffer (25 mM Tris base, 1.9 M glycine, 70% water and 20% methanol). The transfer unit (BioRad) was assembled and run at 100V for 1 hour. Transfer of proteins to the nitrocellulose membrane (Protran, Midwest Laboratories) was then confirmed using Ponceau S dye (0.1% Ponceau S and 1% acetic acid). The membrane was then incubated in 10 ml Blotto (7% dry milk, 9.98 mM Tris base, pH 8.0, 1.5 M NaCl, 0.1% Tween 20 and 0.1% Antifoam A) for 20 minutes at room temperature with gentle agitation. The primary anti-T7 monoclonal mouse antibody (Novagen) was added directly to the Blotto and incubated 1 hour at room temperature with gentle agitation. The membrane was then rinsed three times for two minutes each in 1X Tris Saline (9.98 mM Tris base, pH8.0 and 1.5 M NaCl). Ten milliliters of fresh Blotto containing 1:2500 dilution of a secondary antibody (a goat anti-mouse antibody) conjugated to horse radish peroxidase (Kirkegaard and Perry Laboratories) was incubated with the membrane for an hour. Following incubation with the secondary antibody, the blot was washed three times for five minutes each in 1X Tris Saline. Two milliliters of horse radish peroxidase substrate True Blue (Kirkegaard and Perry Laboratories) was then added directly to the drained blot to detect antibody binding. Foto/Analyst Archiver (Fotodyne) was used to photograph the blot after detection.
Activity Assays
Protein was expressed from 10 ml of BL21(DE3) cells (OD600=0.5) for two hours, isolated using the protocol above and resuspended in a small volume of IDH assay buffer (0.2 M Tris, 1 mM MgCl2, pH 8.0). Reaction solutions were mixed in the 200 ul capacity microwells of a 96-well plate, and analyzed using a multi-well plate spectrophotometer (BioRad Model 3550-UV Microplate Reader). Porcine heart NADP+-dependent isocitrate dehydrogenase (Sigma, product number I-2002) was used as a positive control to test reaction solutions. The positive control reaction solution consisted of 1:500 dilution of porcine heart IDH, 1.15 mM DL-isocitrate, 720 M NADP+ in assay buffer. Experimental reaction solutions for IDP2 proteins (forward and reverse) contained an aliquot of protein extract in the same reaction conditions as the porcine heart enzyme (ie,1.15 mM DL-isocitrate, 720 M NADP+, 0.2 M Tris, 1 mM MgCl2, pH 8.0.) Negative controls for porcine heart, forward and reverse products contained only enzyme/homogenate and NADP+ in assay buffer, to control for turbidity of enzyme extracts and spontaneous reduction of NADP+ to NADPH. For each reaction receiving DL-isocitrate, this reagent was added simultaneously to each well using a multi-tip pipettor just before insertion into the spectrophotometer. The OD340 of each reaction solution was assessed every 30 seconds for a total of 5 minutes to measure the appearance of NADPH. Activity was estimated by linear regression.
Polymerase Chain Reaction
The first objective of the project was to amplify the IDP2
gene from yeast genomic DNA. The published IDP2 sequence (Loftus
et al., 1994) was used to create primers. The primers were
designed to amplify the 1.2 kb IDP2 gene sequence, adding
a BamHI site just before the start codon, and a HindIII site just
after the stop codon. Genomic DNA was purified for use as a template
in PCR (Kaiser et al.1994). After the PCR reaction was
performed, the product was analyzed by agarose gel electrophoresis
to confirm amplification of the expected 1.2 kb IDP2 gene
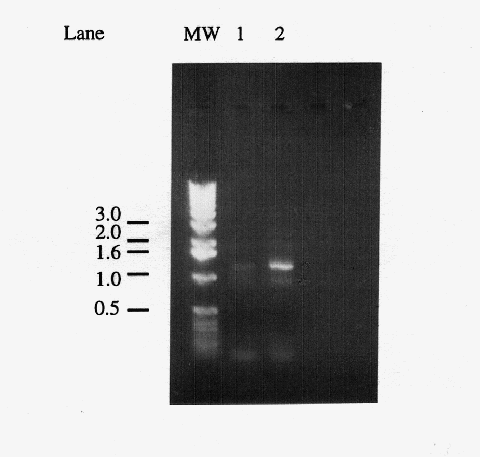
(Figure 1). The gel revealed that the PCR amplified a band of
the expected size, confirming that the reaction was successful.
A small cube of agarose surrounding the 1.2 kb band was excised,
and the DNA was electroeluted from the gel for use in ligation
reactions.
Figure 1. Comparison of primary and secondary PCR products. Five microliters of "primary" PCR product from a reaction using yeast genomic DNA as template (lane 1) and a "secondary" PCR product which used 1 l of a primary PCR product as template (lane 2). The 1.2 kb band corresponds to the amplified IDP2 gene, the light band at ~0.9 kb is a non-specific amplification product, and the diffuse bands below the 0.5 kb marker are most likely primer dimers. Markers have been labeled in kb.
Analysis of pCR2.1-IDP2 clones
The second stage of the project was to clone the IDP2
PCR product directly into pCR2.1, the TA Cloning® (Invitrogen)
plasmid. The TA Cloning system takes advantage of the adenine
overhangs added to the PCR product's 3' ends by Taq polymerase.
At pCR2.1's pre-cut insertion sites, thymidine overhangs have
been added to the 3' ends, which allow direct ligation of PCR
products. The pCR2.1 plasmid is equipped for blue-white screening,
such that colonies containing plasmids without inserts appear
blue. When TOP10F' cells were transformed with the pCR2.1-IDP2
ligation product, approximately 45% of colonies were white, suggesting
clones with inserts. Twelve white colonies were chosen for restriction
digestion analysis with BamHI and HindIII. Digestion products
were visualized using gel electrophoresis (Figure 2). Of the twelve
colonies, two appeared to contain inserts of 1.2 kb. Plasmid DNA
from the colonies containing a 1.2 kb band was digested with BamHI,
HindIII, and EcoRI to confirm the band as IDP2, which contains
an internal EcoRI site 359 bp from the start codon. Gel electrophoresis
revealed that in both colonies the 1.2 kb band was digested into
~400 and ~800 bp bands, confirming the 1.2 kb band's identity
as IDP2. A large scale preparation of pCR2.1-IDP2
plasmid DNA was isolated and digested with BamHI. Then IDP2
was gel purified for ligation into the pET5a expression vector.
Digestion of pCR2.1-IDP2 with BamHI excizes the entire
IDP2 gene sequence in tandem with 36 bp of the pCR2.1 polylinker
at its 3' end. This relationship is maintained throughout cloning
and expression in pET5a.
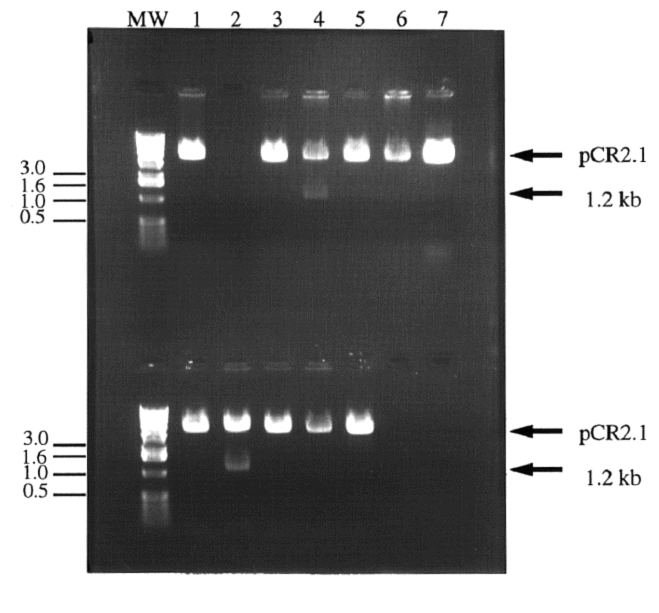
Figure 2. Digestion of colony plasmid DNA in search
of cloned IDP2 PCR product. Twelve white colonies
were chosen for screening. The colonies' plasmid DNA was isolated
and digested with BamHI and HindIII to release the 1.2 kb IDP2
PCR product. Most colonies contained pCR2.1 without the IDP2
insert (1a, 3a, 5a,6a, 7a, and 1b, 3b, 4b, 5b and 6b) or nothing
(2a). Two clones appeared to contain the IDP2 insert (4a
and 2b). Lanes 7b and 8b contained no sample. Molecular weight
markers have been indicated in kb.
Analysis of pET5a-IDP2 clones
In order to produce IDP2 protein for analysis, it was necessary
to clone the IDP2 gene sequence in the expression vector
pET5a. The BamHI digested and purified IDP2 sequence was
ligated into BamHI-digested pET5a. Seven colonies appeared after
JM109 cells were transformed with the pET5a-IDP2 ligation
product. Because the IDP2 gene was ligated into pET5a using
only a single restriction enzyme, two different ligation products
could result (Figure 3). Some colonies could contain IDP2
ligated into pET5a in the forward orientation, for transcription
of a sense RNA (pET5a-IDP2F). Others could contain IDP2
in the reverse orientation (pET5a-IDP2R) resulting in antisense
RNA. The seven colonies which appeared were screened using an
EcoRI digestion designed to determine if the colonies contained
IDP2 and in what orientation (Figure 4). When digested
with EcoRI, forward constructs released a ~950 bp fragment. Reverse
constructs released two visible bands, the ~950 bp band and a
~350 bp band. One colony contained the reverse construct pET5a-IDP2R,
while four colonies contained the forward construct, pET5a-IDP2F.
Plasmid DNA was isolated from one colony containing pET5a-IDP2F
and one containing pET5a-IDP2R for use in transforming
BL21(DE3) expression cells.
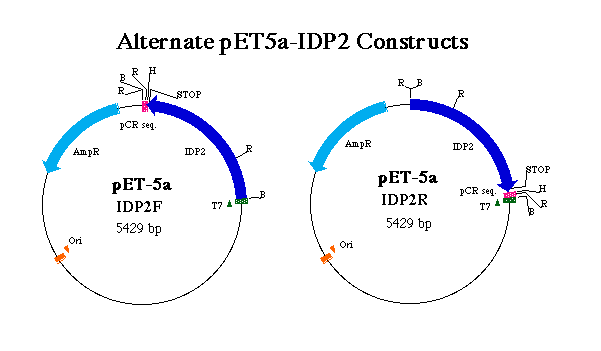
Figure 3. Representation of forward and reverse pET5a-IDP2 constructs. T7 indicates the T7 tag region which initates transcription of the T7 tag in tandem wit the IDP2 coding region. pCR denotes sequence digested from pCR2.1 in tandem with the IDP2 gene. AmpR indicates the ampicillin resistance gene and Ori the origin of replication. R, B and H denote EcoRI, BamHI and HindIII restriction sites respectively. Notice the positions of stop codons and the result of an EcoRI digest on each construct.
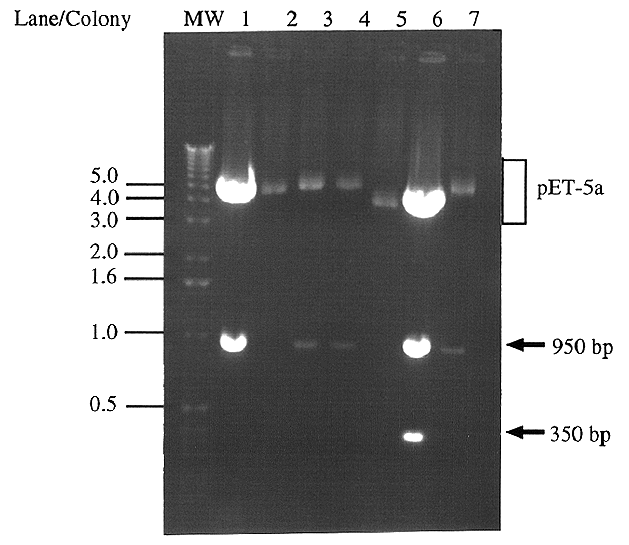
Figure 4. Digestion of pET5a clones' plasmid DNA
for presence and orientation of IDP2 All seven colonies
which appeared after JM109 cells were transformed with the pET5a-IDP2
ligation product were screened using an EcoRI digestion. Forward
clones (pET5a-IDP2F) digest to give a ~950 bp band (lanes
1,3,4 and 7), whereas reverse clones (pET5a-IDP2R) give
a ~950 bp and a ~350 bp band (lane 6). The remaining two colonies
did not contain an insert (lanes 2 and 5). Molecular weight markers
are indicated in kb.
Immunoblot Analysis
The T7 promoter region of pET5a initiates transcription upstream
of the insertion point of the added sequence. Translation of the
resulting RNA gives a protein with the 12 amino acid T7 epitope-tag
on the N-terminus. In pET5a-IDP2F, translation of the full
length sense RNA transcript results in an IDP2 protein with the
T7 tag on the N-terminus. Expression from pET5a-IDP2R results
in a 36 amino-acid chain including the 12 amino acids of the T7
tag at the N-terminus and 24 amino acids of the antisense transcripts
of the pCR2.1 polylinker and IDP2 (Figure 5). Induction
of expression and purification of the resulting protein was performed
in parallel with cells containing pET5a-IDP2F and cells
containing pET5a-IDP2R. Protein from cells containing pET5a-IDP2F
was used as a negative control in immunoblot analysis.
An immunoblot was performed to confirm that the protein expressed
in pET5a-IDP2F was of the correct molecular weight (46,535
kDa), that no visible protein was expressed in the pET5a-IDP2R
clones, and to compare DNA removal methods (Figure 6). The blot
provided evidence that the expressed protein is of the expected
size (47,863 Da, Figure 7), no detectable protein is produced
by the reverse construct, and that all three methods of DNA removal
(short, long DNAse treatments, and mechanical shearing) are viable.
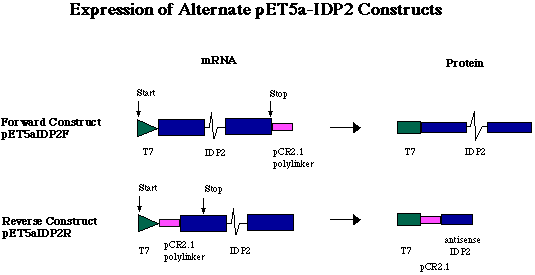
Figure 5. Expression of alternate pET5a-IDP2 constructs. Because a short sequence of polylinker was digested from pCR2.1 in tandem with the 3' end of the gene, the pCR2.1 fragment is transcribed in tandem with the IDP2 gene. In the forward construct (pET5a-IDP2F), a sense RNA is transcribed carrying the 12 codons of the T7 tag on the 5' end of the IDP2 transcript, and the pCR2.1 polylinker fragment on the 3' end. The pCR2.1 fragment is not translated, however, because IDP2 stop codon ends translation before reaching the pCR2.1 fragment. In the reverse construct (pET5a-IDP2R), the IDP2-pCR2.1 fragment was ligated into the pET5a plasmid in the opposite orientation. This ligation event placed the antisense DNA of pCR2.1 and IDP2 directly downstream of the T7 promoter and tag. The antisense RNA of pCR2.1 and IDP2 is translated into a very short amino acid chain (36 aa, approximately 4.0 kDa).
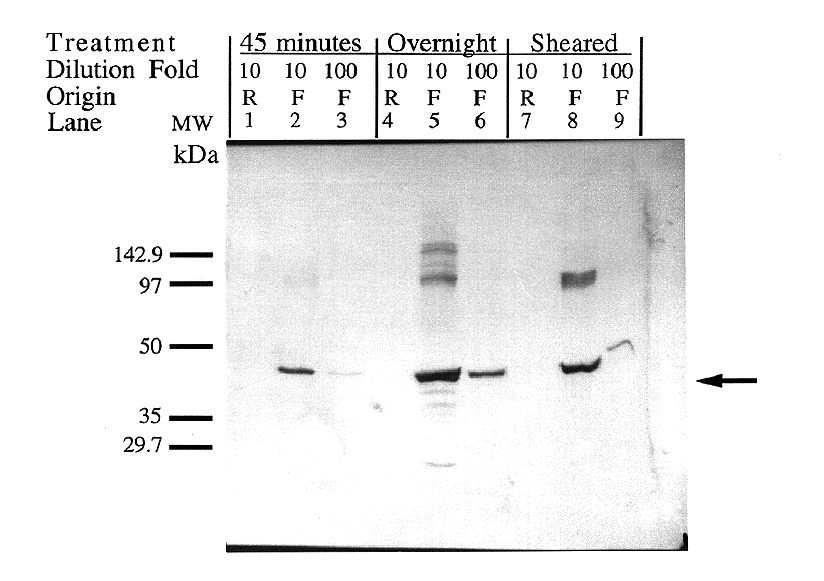
Figure 6. Verification of protein size and comparison
of DNA degradation methods. Expression was induced in both
forward and reverse clones for one hour using 0.4 mM IPTG. Crude
lysate protein was treated in three different ways to degrade
contaminate DNA. Lanes 1-3 contain samples treated with 100 g/ml
DNase for 45 minutes. Lanes 4-6 contain samples treated with 10
g/ml DNAse for 19 hours. Lanes 7-9 contain samples whose DNA was
degraded by means of mechanical shearing by 50 strokes of the
entire lysate volume through a 27.5-gauge needle. The first lane
in each set (1,4,7) contains a 1:10 dilution of the truncated
antisense expression product (Reverse orientation). The
second (2, 5, 8) and third lanes (3, 6, 9) in each set contain
a 1:10 and 1:100 dilution, respectively, of the full-length sense
expression product (Forward orientation). The IDP2 protein
was calculated to be approximately 47 kDa, comparable to the expected
size of 50 kDa.
Activity Assay
Protein products from both forward and reverse constructs
of pET5a-IDP2 were tested for NADP+-dependent isocitrate dehydrogenase
activity. Through comparison to pig heart NADP+-dependent isocitrate
dehydrogenase, the protein isolated from cells containing the
forward T7-IDP2 construct was estimated to have 5.44e-3 units
of IDH activity /ml culture induced at OD600= 0.5 for 2 hours.
Protein isolated from cells containing the reverse T7-IDP2 protein
exhibited negligible activity, suggesting that the activity of
protein of cells containing the forward construct is a result
of the recombinant T7-IDP2 protein itself and not E.coli
IDH (Figure 7).
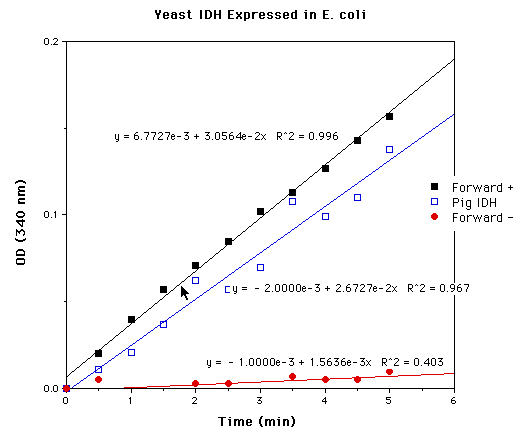
Figure 7: Yeast IDH expressed in E.coli. Forward + assay contained all reagents necessary for reaction; Forward- assay contained all reagents except isocitrate. Pig heart assay contained 0.0392 mg of IDH, constituting 0.01188 units of IDH activity. Comparison of the slopes of the pig IDH and Forward + assays allows estimation of 0.0136 activity units in the Forward + assay. The Forward+ assay contained protein homogenate isolated from approximately 2.5 ml of E .coli cells induced at OD600 = 0.5 for 2 hours.
This course integrates well with molecular biology and biotechnology
classes, as different types of analyses can highlight facets of
the curriculum. Having a concrete project associated with such
courses effectively involves students in thinking about experimental
design and what occurs at the molecular level as they work. Alternatively,
the course could also be used independently to teach laboratory
methods.
Valuable activities can be added easily during extra laboratory
sessions or when long incubation periods leave excess lab-time.
Students can be involved in preliminary work, such as obtaining
the IDP2 cDNA sequence from a yeast genome databank (Campbell,
1997a) or NIH Genbank (Campbell, 1997b) and/or preparing the PCR
primers and solutions. DNA and protein analysis can be introduced
to students during the course, using software such as MacDNAsis
and/or CaseIt. MacDNAsis allows users to perform both DNA and
peptide analysis, including searches for open reading frames or
restriction sites, alignment of sequences, and prediction of peptides'
primary and secondary characteristics. CaseIt provides a method
to simulate or predict the results of DNA manipulation procedures
such as PCR, restriction digestions, agarose gels, and Southern
Blots. After the basic procedures have been completed, further
work can be done to analyze the recombinant IDP2 DNA, RNA or protein.
Southern and Northern blots can be performed using nucleic acids
isolated from transformed BL21(DE3) cells. Alternatively, several
species' genomic DNA can also be isolated in the lab or obtained
from major biotechnology companies for use in a multi-species
Southern blot (zooblot).
This zooblot can allow students to compare interspecific differences
between IDH genes and the importance of stringency conditions
during probing.. In a similar way, Northern blots can be performed
using RNA. The recombinant IDP2 gene can be used as a probe in
such procedures.
Analysis of IDP2 activity is especially pertinent as the active
site of IDP2 has not yet been localized. At least one study has
shown that the N-terminus of tobacco cytosolic NADP+-dependent
isocitrate dehydrogenase is important to activity or adhesion
to other subunits. Allowing students to view RasMol
images of IDH molecules may aid students in making educated
guesses about whether or not the T7-IDP2 recombinant protein is
active. Activity can be determined using spectrophotometric activity
assays (Williamson, et al., 1980) or a native polyacrylamide
gel tested for activity (Davis, 1964).
Campbell, M.A. (1997a). How to search genbank for the sequence of your favorite protein (YFP). Davidson College Biology Department. <http://bio.davidson.edu/Biology/Courses/Molbio/searchYFP.html> Accessed 1998 Mar 23
Campbell, M.A. (1997b) How to search and retrieve from the NIH genbank. Davidson College Biology Department. <http://bio.davidson.edu/Biology/Courses/Molbio/NIHsearch.htm> Accessed 1998 Mar 23
Campbell, M.A. (1997c) IDH as a Model Enzyme for teaching biology. Davidson College Biology Department <http://bio.davidson.edu/Biology/RasMol/IHD1AI3.pdb> Accessed 1998 May 4
Davis, B.J. (1964). Disc Electrophoresis II: Method and application to human serum proteins. Annals of the New York Academy of Science. 121:404-427
Deutscher, Murray P. (Ed.) (1990). Guide to Protein Purification. Methods in Enzymology. Volume 182. San Diego: Academic Press Inc.
Galvez, S., Hodges, M., Bismuth, E., Samson, I., Teller, S., and Gadal, P. (1995). Purification and characterization of a fully active recombinant tobacco cytosolic NADP+-dependent isocitrate dehydrogenase in Escherichia coli: Evidence for a role for the N-terminal region in enzyme activity. Archives of Biochemistry and Biophysics. 323(1): 164-168.
Kaiser, C., Michaelis, S., Mitchell, A. (1994). Methods in Yeast Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press
Loftus, T.M., Hall, L.V., Anderson, S.L. and McAlister-Henn,
L. (1994). Isolation and
characterization of the yeast gene encoding cytosolic NADP+-specific
isocitrate
dehydrogenase. Biochemistry. 33: 9661-9667.
Sambrook, J., Fritsch, E.F., and Maniatis, T. (1989). Molecular Cloning. 2nd ed. USA: Cold Spring Harbour Laboratory Press.
Williamson, J.H., Krochko, D., and Bentley, M. (1980). Properties of Drosophila NADP+- isocitrate dehydrogenase purified on Procion Brilliant Blue-Sepharose-4B. Comparative Biochemical Physiology 65B:339-343
We would like to thank Dr. John L. Woolford for providing us with yeast cells, glass beads, and protocols for storing and obtaining genomic DNA from the yeast cells. I would also like to thank Dr. John Williamson and Ms. Allison Coble, who were consulted for technical advice, critical readings, and encouragement. Dr. Malcolm Campbell was invaluable partner in the design, performance, and presentation of this project.