This web page was produced as an assignment for an undergraduate course at Davidson College
Overview |
Journal Articles |
Popular Press |
Conclusion |
|---|
ABCA3 Gene & Infant Lung Disease
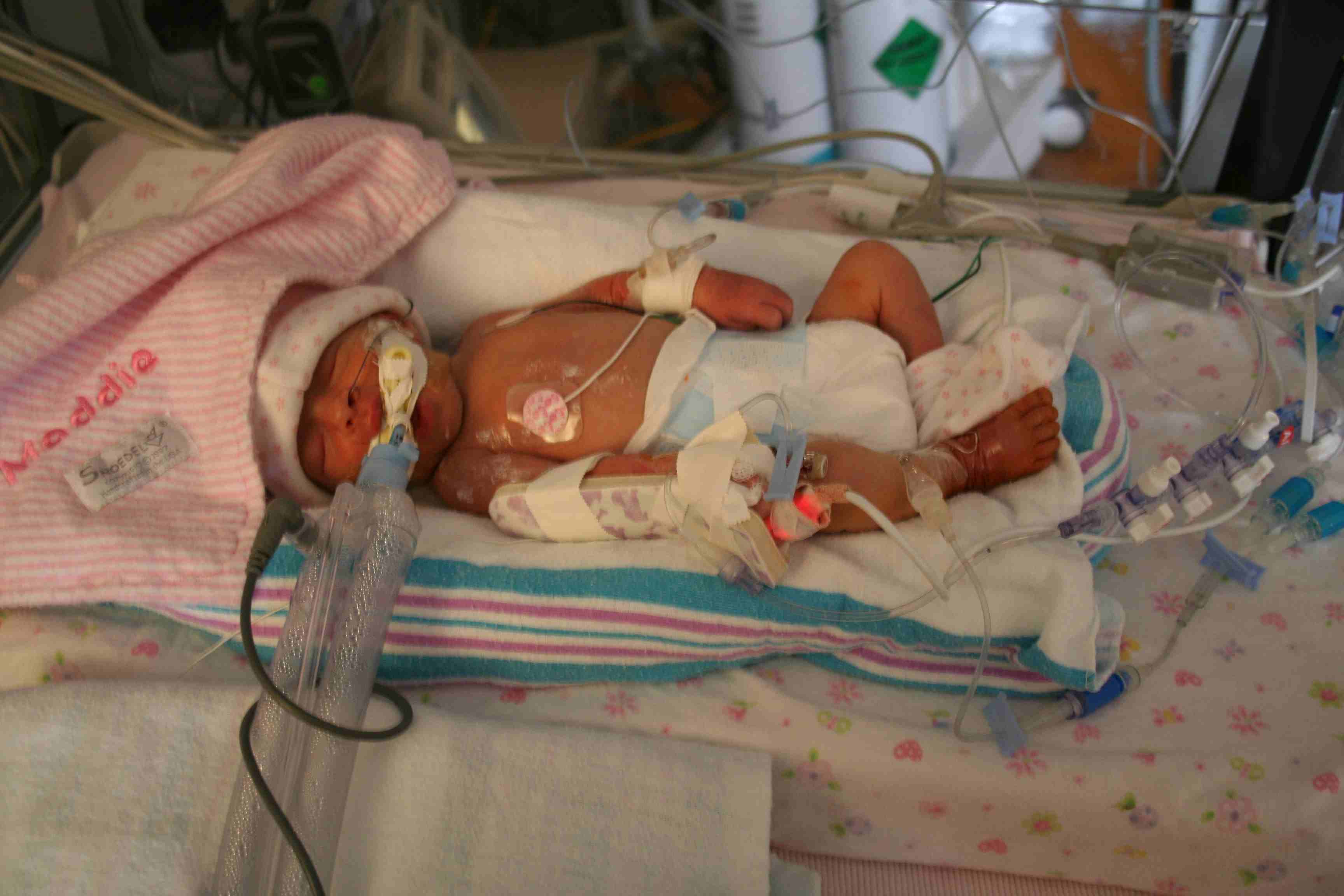
Picture of ABCA3 Surfactant protein deficient infant - Maddie. Image taken after birth. Thanks to Schmit's Science Synopsis. Permission Pending
Purpose:
Lung diseases come in various forms. Some originate from harsh environments, while others spawn from the very material that makes us who we are. Infant lung disease associated with surfactant dysfunction is one such case that studies utilizing genetics has attempted to understand. By analyzing DNA from numerous patients, researchers and doctors have narrowed their search down to a few key mutations found in the genes encoding the surfactant proteins B and C (SP-B and SP-C) and the phospholipid transporter, ABCA3 (Wert, 2009). Problem solved, right? Wrong! Issues correlating genetic variations and mutations with phenotypic consequences are becoming more and more complicated. As such, a new emerging field integrating various aspects of genetics with large-scale systems biology attempts to unravel these new age problems. This groundbreaking field, known as genomics, presents opportunities of understanding far beyond that of our current knowledge; flirting with breakthroughs that could change the way biology and medicine is seen today.
The rule of: one gene, one function, no longer remains true in all cases. Instead, a new perspective is required. This webpage will attempt to shed light on this new perspective of genomics by comparing and contrasting scientific publications and popular press articles addressing the same issue. Specifically, this discussion will highlight how findings are presented for the two types of articles and what aspects are best conveyed by each one respectively.
Background:
The case study under consideration here involved the identification of an unknown cause of severe respiratory disease in infants as part of a study to distinguish inherited abnormalities of surfactant metabolism. Previous studies have documented genetic disorders disrupting normal surfactant metabolism as causes of respiratory disease in newborn and developing infants. This disruption of surfactant in the lungs occurs primarily during development and has since been linked with mutations in the genes encoding the surfactant proteins B and C (SP-B and SP-C) and the phospholipid transporter, ABCA3 (Wert, 2009). Before comparing each articles standpoint and presented findings, let us first define some terms essential to understanding surfactant dysfunction and examine its associated phenotypic expression as lung disease.
Pulmonary surfactant, a mixture of lipids and proteins, lowers surface tension in alveolar type II cells thus preventing end-expiratory atelectasis or lung collapse in laymen’s terms. Surfactant is primarily stored in alveolar type II cells, specifically in lysosomal-derived intracellular inclusions known as lamellar bodies (LB). From here, surfactant is secreted into the alveoli by exocytosis (Shulenin et al., 2004). Surfactant proteins B and C are small 79-amino-acid and 35-amino-acid proteins respectively, that assist in the absorption of the surfactant phospholipid film to the air-liquid interface, thereby contributing to the maintenance of the surface tension-reducing characteristics of surfactant.
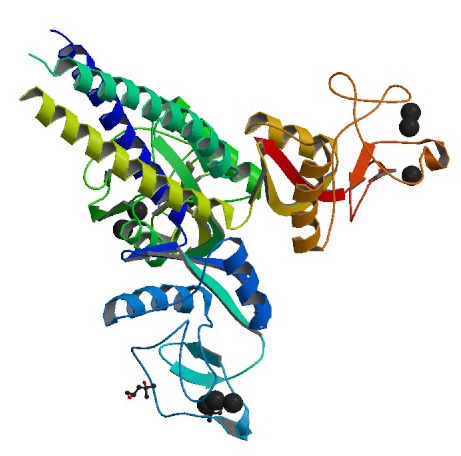
Crystal structure of head and neck domain of Surfactant protein D (SP-D), a closely related family member to SP-B and SP-C. While no mutations in this gene has been identified to date,
SP-D-deficient mice demonstrated defects in regulation pathways of secreted surfacnt phospholipids by alveolar type II cells (Wert, 2009). Image obtained from PDB.
ATP-binding cassette A3 (ABCA3), however, belongs to a superfamily of genes that encode lipid transport proteins in the body. ABAC3, in particular, encodes a membrane transport protein in humans that has been linked with surfactant phospholipid transport across intracellular membranes. This protein, expressed in various types of tissues including lung, brain, and kidney, has been associated with lung disease in infants (Besnard et al., 2010). ATP-binding cassette (ABC) genes are divided into a set of seven distinctive subfamilies, ABCA3 being a member of the ABC1 subfamily, is found exclusively in multicellular eukaryotes (NCBI). This 190-kDa protein is comprised of 1,704-amino acids and is located on chromosome 16 (16p13.3) in Homo sapiens (Besnard et al., 2010).
In addition to being expressed in the brain and kidneys, ABAC3 is highly expressed in the limiting membrane of lamellar bodies (LB). This expression is especially abundant at the membrane of LBs in type II alveolar epithelium cells (Bullard et al., 2005). Because of ABCA3s localization and its numerous interactions with SP-B and SP-C, as well as the transportation of surfactant in general, ABCA3 appears to play an important role in the development of lamellar bodies and surfactant production.
References
Bullard, Janine E., Susan E. Wert, Jeffery A. Whitsett, Michael Dean, and Lawrence M. Nogee. "ABCA3 Mutations Associated with Pediatric Interstitial Lung Disease." American Journal of Respiratory and Critical Care Medicine 172.8 (2005): 1026-031. American Journal of Respiratory and Critical Care Medicine. American Thoracic Society, 23 June 2005. Paper Link.
Besnard, Valerie, Yohei Matsuzaki, Jean Clark, Yan Xu, Susan E. Wert, Machiko Ikegami, Mildred T. Stahlman, Timothy E. Weaver, Alan N. Hunt, Anothony D. Postle, and Jeffery A. Whitsett. "Conditional Deletion of Abca3 in Alveolar Type II Cells Alters Surfactant Homeostasis in Newborn and Adult Mice." American Journal of Physiology. Lung Cellular and Molecular Physiology 298 (2010). Lung Cellular and Mol. American Physiological Society, 26 Feb. 2010. Paper Link.
Shulenin, Sergey, Lawrence M. Nogee, Tarmo Annilo, Susan E. Wert, Jeffrey A. Whitsett, and Michael Dean. "ABCA3 Gene Mutations in Newborns with Fatal Surfactant Deficiency." New England Journal of Medicine 350.13 (2004): 1296-303. Paper Link.
Wert, Susan E., Jeffrey A. Whitsett, and Lawrence M. Nogee. "Genetic Disorder of Surfactant Dysfunction." Pediatric and Developmental Pathology: The Official Journal of the Society for Pediatric Pathology and the Paediatric Pathology Society 12.4 (2009). NCBI: U.S. National Library of Medicine. National Institutes of Health. Society for Pediatric Pathology, July-Aug. 2009. Paper Link.
William's Home Page
Genomics Page
Biology Home Page
Email: William Green for questions or comments.
© Copyright 2011 Department of Biology, Davidson College, Davidson, NC 28035