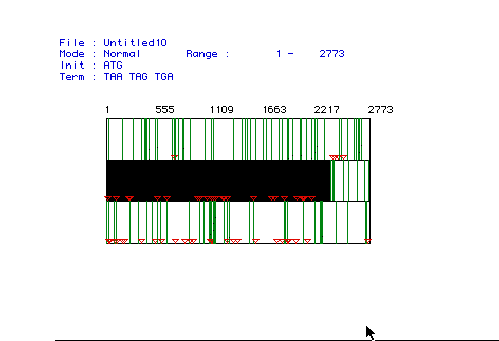
I used the MacDNAsis program to obtain:
Figure 1: MacDNAsis was used to determine the largest ORF for the cDNA of Canis familiaris. The three rows show the three possible reading frames of the cDNA sequence (starting at the first nucleotide, the second nucleotide, or the third nucleotide). The red triangles and the green rectangles represent start codons and stop codons, respectively. The large black box indicates the largest ORF from nucleotide 23 to nucleotide 2371.
Predicted Molecular Weight:
The predicted molecular weight of phosphofructokinase was determined
by translating the protein DNA into amino acids using the MacDNAsis program.
The number of amino acids played an important role in predicting the molecular
weight at 85556.55 daltons.
Hydropathy Plot:
A hydropathy plot was used to determine whether phosphofructokinase
is an integral membrane protein or not. The Kyte and Doolittle plot
shows the hydrophobicity of the protein, with the positive values indicating
more hydrophobic regions. Kyte and Doolittle have proposed that any
protein that contains a portion with a reading of over +1.8 is a good candidate
for an integral membrane protein.
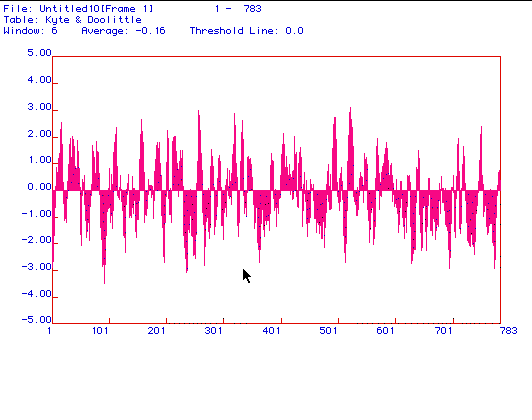
Figure 2: The hydropathy plot generated under the parameters
of Kyte and Doolittle using MacDNAsis indicates whether a protein is an
integral membrane protein. Positive numbers signify hydrophobic regions
with more hydrophobic regions having higher values. The determined
value for a suggested integral membrane protein is +1.8.
Several of the peaks in the figure above have a hydrophobicity that is greater than +1.8 (at least twelve points exceed 2.0), suggesting that phosphofructokinase is an integral membrane protein.
Antigenicity Plot:
The Hopp and Woods plot is used to determine what portion of the protein
is more antigenic and can be used to make a peptide. This peptide
could then be used to make a monoclonal antibody against the peptide.
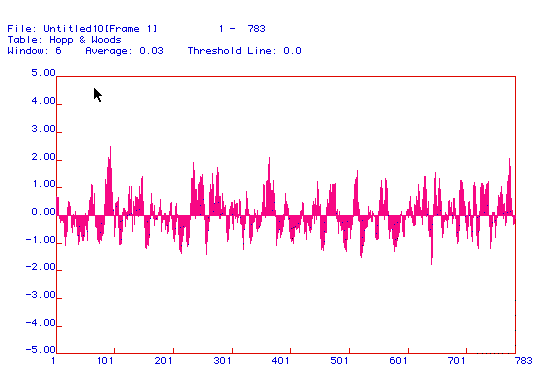
Figure 3: The Hopp and Woods antigenicity plot shows
the hydrophilic regions (as opposed to the hydrophobic regions displayed
in the Kyte and Doolittle plot) of the protein.
Positive values indicate more antigenic areas of phosphofructokinase.
These regions are directed away from the hydrophobic phospholipid bilayer
of the membrane and can be used to generate peptides against which monoclonal
antibodies can be made. As seen in the above plot, there are many
hydrophilic areas, especially at 101, 250, and 375.
Predicted Secondary Structure:
The secondary structure of phosphofructokinase can be predicted using
the Chou, Fasman, and Rose plot which takes into account amino acid interactions,
including through side chains and hydrogen bonding, and the hydrophobicity
of individual amino acids.
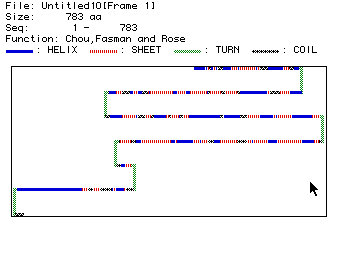
Figure 4: The Chou, Fasman, and Rose plot shows the predicted
secondary structure of phosphofructokinase. Types of structure are
displayed above the graphic.
As pictured in Figure 4, the secondary structure of phosphofructokinase includes
six turns and many helices, sheets, and coils. The secondary structure
can also be view in 3D with the Rasmol Image.
This smaller portion of the protein includes 44 helices and 44 strands.
Multiple Sequence Alignment:
This technique compares the amino acid sequences of five organisms: Canis familiaris
(DOG), Mus musculus (MOUSE),
Drosophila melanogaster (FLY),
Homo sapiens (HUMAN),
and Caenorhabditis elegans (PLANT).

Figure 5: The multiple sequence alignment of all five
proteins displays the homology between the amino acid sequences of the
different organisms. Black boxes with yellow letters indicate conserved
amino acids.
The short section of the sequence alignment shown in Figure 5 suggests that this is a highly conserved sequence in all five organisms. The high degree of homology indicates that these sequences have not diverged much over time.
Phylogenetic Tree:
A phylogenetic tree, using the Higgins method, exhibits the degree
of amino acid conservation over time of the amino acid sequences of the
five organisms.
To see the cDNA sequences of these organism, click on the following links:
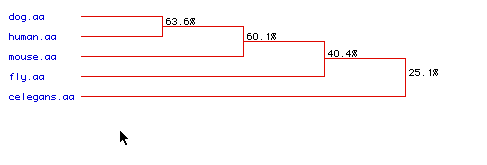
The dog and human amino acid sequences are most conserved, followed closely by the mouse amino acid sequence. This result was also obtained in Figure 5 in the multiple sequence alignment. The least conserved amino acid sequence was C. elegans, but even the 25.1% conservation is fairly high considering that the placement of the same amino acid in the same position is only 5%, and that is for each single amino acid. To obtain an entire sequence with the same amino acids in the same order would be even more unlikely.
e-mail any questions, comments, or concerns to me at: